Genetická porucha - Genetic disorder
| Genetická porucha | |
|---|---|
 | |
| Chlapec s Downův syndrom, jedna z nejčastějších genetických poruch | |
| Specialita | Lékařská genetika |
A genetická porucha je zdravotní problém způsobený jednou nebo více abnormalitami v genom. Může to být způsobeno a mutace v jednom gen (monogenní) nebo více genů (polygenní) nebo a chromozomální abnormalita. Ačkoli polygenní poruchy jsou nejčastější, tento termín se většinou používá při diskusi o poruchách s jedinou genetickou příčinou, buď v genu nebo chromozóm.[1][2] Odpovědná mutace se může objevit spontánně dříve embryonální vývoj (A de novo mutace), nebo může být zdědil od dvou rodičů, kteří jsou nositeli vadného genu (autozomálně recesivní dědictví) nebo od rodiče s poruchou (autosomálně dominantní dědictví). Některé poruchy jsou způsobeny mutací na X chromozom a mít X-vázaný dědictví. Velmi málo poruch se dědí na Y chromozom nebo mitochondriální DNA.[3]
Existuje více než 6 000 známých genetických poruch,[4] a v lékařské literatuře jsou neustále popisovány nové genetické poruchy.[5] Asi 1 z 50 lidí je postiženo známou poruchou jednoho genu, zatímco přibližně 1 z 263 je postiženo a chromozomální porucha.[6] Asi 65% lidí má nějaké zdravotní problémy v důsledku vrozených genetických mutací.[6] Vzhledem k významně velkému počtu genetických poruch je přibližně 1 z 21 lidí postiženo genetickou poruchou klasifikovanou jako „vzácný „(obvykle definováno jako postihující méně než 1 z 2 000 lidí). Většina genetických poruch je sama o sobě vzácná.[5][7]
Rakoviny jsou způsobeny genetickými mutacemi, ale jsou obecně vynechány, pokud jde o genetické poruchy, protože většina není dědičná (i když predispozice a rakovinové syndromy existovat).[8]
Single-gen
| Prevalence poruchy (přibližná) | |
|---|---|
| Autozomálně dominantní | |
| Familiární hypercholesterolemie | 1 z 500[9] |
| Polycystické onemocnění ledvin | 1 ze 750[10] |
| Neurofibromatóza typu I | 1 z 2 500[11] |
| Dědičná sférocytóza | 1 z 5 000 |
| Marfanův syndrom | 1 ze 4 000[12] |
| Huntingtonova choroba | 1 z 15 000[13] |
| Autosomálně recesivní | |
| Srpkovitá anémie | 1 ze 625[14] |
| Cystická fibróza | 1 z 2 000 |
| Tay – Sachsova choroba | 1 z 3 000 |
| Fenylketonurie | 1 z 12 000 |
| Mukopolysacharidózy | 1 z 25 000 |
| Nedostatek lysozomální kyselé lipázy | 1 ze 40 000 |
| Nemoci z ukládání glykogenu | 1 z 50 000 |
| Galaktosemie | 1 z 57 000 |
| X-vázaný | |
| Duchennova svalová dystrofie | 1 z 5 000 |
| Hemofilie | 1 z 10 000 |
| Hodnoty jsou pro živě narozené děti | |
A porucha jednoho genu (nebo monogenní porucha) je výsledkem singlu zmutovaný gen. Poruchy jednoho genu lze přenést na následující generace několika způsoby. Genomický otisk a uniparental disomy, však může ovlivnit vzory dědičnosti. Rozdíly mezi recesivní a dominantní typy nejsou „tvrdé a rychlé“, i když rozdíly mezi nimi autosomální a X-vázaný typy jsou (protože poslední typy se rozlišují čistě na základě chromozomální polohy genu). Například běžná forma nanismus, achondroplázie, je obvykle považována za dominantní poruchu, ale děti se dvěma geny pro achondroplázii mají těžkou a obvykle smrtelnou kosterní poruchu, kterou lze považovat za nositele achondroplazmy. Srpkovitá anémie je také považován za recesivní stav, ale heterozygotní nosiče mají zvýšenou odolnost vůči malárie v raném dětství, které lze popsat jako související dominantní stav.[15] Pokud si pár, kde jeden z partnerů nebo oba trpí nebo jsou nositeli poruchy jednoho genu, přeje mít dítě, může tak učinit prostřednictvím in vitro oplodnění, které umožňuje předimplantační genetickou diagnostiku ke kontrole, zda má embryo genetickou poruchu.[16]
Většina vrozená metabolické poruchy známé jako vrozené poruchy metabolismu výsledkem defektů jednoho genu. Mnoho takových defektů s jedním genem může snížit kondici postižených lidí, a proto jsou v populaci přítomny s nižší frekvencí ve srovnání s očekáváním na základě jednoduchých pravděpodobnostních výpočtů.[17]
Autozomálně dominantní
Pouze jedna mutovaná kopie genu bude nezbytná k tomu, aby byla osoba postižena autozomálně dominantní poruchou. Každá postižená osoba má obvykle jednoho postiženého rodiče.[18]:57 Šance, že dítě zdědí mutovaný gen, je 50%. Autosomálně dominantní podmínky se někdy snížily pronikavost, což znamená, že i když je potřeba pouze jedna mutovaná kopie, ne u všech jedinců, kteří zdědí tuto mutaci, se onemocnění vyvine. Příklady tohoto typu poruchy jsou Huntingtonova choroba,[18]:58 neurofibromatóza typu 1, neurofibromatóza typu 2, Marfanův syndrom, dědičná nepolypózní kolorektální rakovina, dědičné mnohočetné exostózy (vysoce pronikavá autosomálně dominantní porucha), tuberózní skleróza, Von Willebrandova choroba, a akutní přerušovaná porfyrie. Vrozené vady se také nazývají vrozené anomálie.
Autosomálně recesivní
Aby mohla být osoba postižena autozomálně recesivní poruchou, musí být mutovány dvě kopie genu. Postižená osoba má obvykle neovlivněné rodiče, kteří každý nosí jednu kopii mutovaného genu a jsou označováni jako genetické nosiče. Každý rodič s defektním genem obvykle nemá příznaky.[19] Dva nepostižení lidé, z nichž každý nese jednu kopii mutovaného genu, mají s každým těhotenstvím 25% riziko, že dítě bude postiženo touto poruchou. Příklady tohoto typu poruchy jsou albinismus, nedostatek acyl-CoA dehydrogenázy se středním řetězcem, cystická fibróza, srpkovitá nemoc, Tay – Sachsova choroba, Niemann – Pickova choroba, spinální svalová atrofie, a Robertsův syndrom. Některé další fenotypy, například mokré vs. suché ušní maz, jsou také určovány autozomálně recesivně.[20][21] Některé autozomálně recesivní poruchy jsou běžné, protože v minulosti nesení jednoho z chybných genů vedlo k mírná ochrana proti infekční nemoci nebo toxin jako tuberkulóza nebo malárie.[22] Mezi takové poruchy patří cystická fibróza,[23] srpkovitá nemoc,[24] fenylketonurie[25] a talasémie.[26]

X-vázaná dominantní
Dominantní poruchy vázané na X jsou způsobeny mutacemi genů na X chromozom. Pouze několik poruch má tento vzor dědičnosti, přičemž hlavním příkladem je X-vázaná hypofosfatemická křivice. Muži a ženy jsou těmito poruchami postiženi, přičemž muži jsou obvykle postiženi více než ženy. Některé dominantní podmínky spojené s X, jako např Rettův syndrom, inkontinentia pigmenti typ 2 a Aicardiho syndrom, jsou obvykle fatální také u mužů in utero nebo krátce po narození, a jsou proto převážně pozorovány u žen. Výjimkou z tohoto zjištění jsou extrémně vzácné případy, kdy chlapci s Klinefelterův syndrom (44 + xxy) také zdědí dominantní stav vázaný na X a vykazuje příznaky podobnější těm ženským, pokud jde o závažnost onemocnění. Šance na přenos dominantní poruchy vázané na X se u mužů a žen liší. Synové muže s dominantní poruchou vázanou na X nebudou ovlivněni (protože dostanou chromozom Y svého otce), ale jeho dcery tento stav zdědí všichni. Žena s dominantní poruchou vázanou na X má 50% šanci mít postižený plod s každým těhotenstvím, i když v případech, jako je inkontinentní pigmenti, jsou životaschopní pouze potomci žen.
X-vázaný recesivní
Recesivní podmínky spojené s X jsou také způsobeny mutacemi genů na X chromozomu. Muži jsou mnohem častěji postiženi než ženy, protože mají pouze jeden chromozom X, který je nezbytný pro přítomnost onemocnění. Šance na přenos poruchy se liší u mužů a žen. Synové muže s X-vázanou recesivní poruchou nebudou ovlivněni (protože dostávají Y chromozom svého otce), ale jeho dcery budou nositelkami jedné kopie mutovaného genu. Žena, která je nositelkou recesivní poruchy vázané na X (XRXr) má 50% šanci mít postižené syny a 50% šanci mít dcery, které jsou nositelkami jedné kopie mutovaného genu. Mezi recesivní stavy vázané na X patří vážná onemocnění hemofilie A, Duchennova svalová dystrofie, a Lesch-Nyhanův syndrom, stejně jako běžné a méně závažné stavy, jako je mužská plešatost a červeno-zelená barvoslepost. Recesivní podmínky vázané na X se někdy mohou u žen projevit kvůli vychýlená X-inaktivace nebo monosomie X (Turnerův syndrom ).
Y-vázaný
Poruchy spojené s Y jsou způsobeny mutacemi na chromozomu Y. Tyto stavy mohou být přenášeny pouze z heterogametického pohlaví (např. Muži) na potomky stejného pohlaví. Jednoduše to znamená, že poruchy spojené s Y u lidí lze přenést pouze z mužů na jejich syny; ženy nikdy nemohou být ovlivněny, protože nemají Y-allosomy.
Poruchy spojené s Y jsou mimořádně vzácné, ale nejznámější příklady obvykle způsobují neplodnost. Reprodukce za takových podmínek je možná pouze obcházením neplodnosti lékařským zákrokem.
Mitochondriální
Tento typ dědičnosti, známý také jako mateřská dědičnost, je nejvzácnější a vztahuje se na 13 genů kódovaných mitochondriální DNA. Protože pouze vaječné buňky přispívají mitochondriemi k vyvíjejícímu se embryu, mohou matky (které jsou postiženy) předat podmínky mitochondriální DNA svým dětem. Příkladem tohoto typu poruchy je Leberova dědičná optická neuropatie.
Je důležité zdůraznit, že drtivá většina z mitochondriální nemoci (zvláště když se příznaky objeví v raném věku) jsou ve skutečnosti způsobeny a nukleární gen defekt, protože mitochondrie jsou většinou vyvíjeny nemitochondriální DNA. Tato onemocnění nejčastěji následují autozomálně recesivní dědičnost.[27]
Multifaktoriální porucha
Genetické poruchy mohou být také složité, multifaktoriální nebo polygenní, což znamená, že jsou pravděpodobně spojeny s účinky více genů v kombinaci s životním stylem a faktory prostředí. Mezi multifaktoriální poruchy patří srdeční choroba a cukrovka. Přestože se komplexní poruchy v rodinách často shlukují, nemají jednoznačný vzor dědičnosti. To ztěžuje určení rizika dědičnosti nebo přenosu těchto poruch u osoby. Složité poruchy je také obtížné studovat a léčit, protože dosud nebyly identifikovány specifické faktory, které způsobují většinu těchto poruch. Studie, jejichž cílem je zjistit příčinu komplexních poruch, mohou ke stanovení použít několik metodických přístupů genotyp –fenotyp sdružení. Jednou z metod je přístup založený na genotypu začíná identifikací genetických variant u pacientů a poté určením souvisejících klinických projevů. To je v protikladu k tradičnějšímu přístupu založenému na fenotypu a může to identifikovat kauzální faktory, které byly dříve zakryty klinickými heterogenita, pronikavost a expresivita.
Na rodokmenu mají polygenní nemoci tendenci „běžet v rodinách“, ale dědičnost nesedí k jednoduchým vzorům jako u Mendelian nemoci. To ale neznamená, že geny nelze nakonec lokalizovat a studovat. Mnoho z nich má také silnou složku životního prostředí (např. krevní tlak ).
- astma
- autoimunitní onemocnění jako roztroušená skleróza
- rakoviny
- ciliopatie
- rozštěp patra
- cukrovka
- srdeční choroba
- hypertenze
- zánětlivé onemocnění střev
- mentální postižení
- porucha nálady
- obezita
- refrakční chyba
- neplodnost
Chromozomální porucha
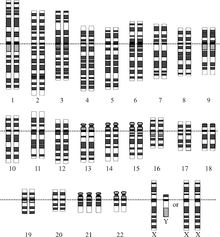
Chromozomální porucha je chybějící, zvláštní nebo nepravidelná část chromozomální DNA. Může to být z atypického počtu chromozomů nebo strukturální abnormality v jednom nebo více chromozomech. Příkladem těchto poruch je trizomie 21 (Downův syndrom ), ve kterém je další kopie chromozomu 21.
Diagnóza
Vzhledem k široké škále známých genetických poruch je diagnóza velmi různorodá a závisí na této poruše. Většina genetických poruch je diagnostikována při narození nebo v raném dětství, nicméně některé, jako např Huntingtonova choroba, může uniknout detekci, dokud se pacient nedostane do dospělosti.
Základní aspekty genetické poruchy spočívají na dědičnosti genetického materiálu. S hloubkou rodinná historie, je možné předvídat možné poruchy u dětí, které směrují zdravotnické pracovníky ke konkrétním testům v závislosti na dané poruše a umožňují rodičům šanci připravit se na možné změny životního stylu, předvídat možnost mrtvé dítě, nebo uvažovat ukončení.[28] Prenatální diagnóza může detekovat přítomnost charakteristických abnormalit ve vývoji plodu prostřednictvím ultrazvuk, nebo detekovat přítomnost charakteristických látek pomocí invazivní postupy které zahrnují zavedení sond nebo jehel do dělohy, například v amniocentéza.[29]
Prognóza
Ne všechny genetické poruchy vedou přímo ke smrti; avšak nejsou známy žádné léky na genetické poruchy. Mnoho genetických poruch ovlivňuje vývojová stadia, jako např Downův syndrom, zatímco jiné mají za následek čistě fyzické příznaky, jako je svalová dystrofie. Jiné poruchy, jako např Huntingtonova choroba, do dospělosti nejeví známky. Během aktivní doby genetické poruchy se pacienti většinou spoléhají na udržení nebo zpomalení degradace kvalita života a udržovat pacienta autonomie. To zahrnuje fyzikální terapie, ovládnutí bolesti, a může zahrnovat výběr alternativní medicína programy.
Léčba

Léčba genetických poruch je pokračující bitva s více než 1 800 genová terapie klinické studie byly dokončeny, probíhají nebo byly schváleny po celém světě.[30] Navzdory tomu se většina možností léčby točí kolem léčby příznaků poruch ve snaze zlepšit pacienta kvalita života.
Genová terapie označuje formu léčby, při které je pacientovi zaveden zdravý gen. To by mělo zmírnit defekt způsobený vadným genem nebo zpomalit progresi onemocnění. Hlavní překážkou bylo dodání genů do příslušné buňky, tkáně a orgánu postiženého poruchou. Jak lze zavést gen do potenciálně bilionů buněk, které nesou defektní kopii? Tato otázka byla překážkou mezi pochopením genetické poruchy a nápravou genetické poruchy.[31]
Epidemiologie
Asi 1 z 50 lidí je postiženo známou poruchou jednoho genu, zatímco přibližně 1 z 263 je postiženo a chromozomální porucha.[6] Přibližně 65% lidí má nějaké zdravotní problémy v důsledku vrozených genetických mutací.[6] Vzhledem k významně velkému počtu genetických poruch je přibližně 1 z 21 lidí postiženo genetickou poruchou klasifikovanou jako „vzácný „(obvykle definováno jako postihující méně než 1 z 2 000 lidí). Většina genetických poruch je sama o sobě vzácná.[5][7] Existuje více než 6 000 známých genetických poruch,[4] a v lékařské literatuře jsou neustále popisovány nové genetické poruchy.[5]
Dějiny
Nejdříve známý genetický stav v a hominid byl ve fosilních druzích Paranthropus robustus, s více než třetinou jednotlivců amelogenesis imperfecta.[32]
Viz také
- FINDbase (databáze Frequency of Inherited Disorders)
- Genetická epidemiologie
- Seznam genetických poruch
- Populační skupiny v biomedicíně
- Mendelianova chyba
Reference
- ^ „Genetické poruchy“. learn.genetics.utah.edu. Citováno 2019-07-01.
- ^ Lvovs, D .; Favorova, O.O .; Favorov, A.V. (2012). „Polygenní přístup ke studiu polygenních chorob“. Acta Naturae. 4 (3): 59–71. doi:10.32607/20758251-2012-4-3-59-71. ISSN 2075-8251. PMC 3491892. PMID 23150804.
- ^ Reference, Genetics Home. „Jaké jsou různé způsoby, jak lze dědit genetický stav?“. Genetická domácí reference. Citováno 2020-01-14.
- ^ A b „OMIM Gene Map Statistics“. www.omim.org. Citováno 2020-01-14.
- ^ A b C d „Orphanet: O vzácných onemocněních“. www.orpha.net. Citováno 2020-01-14.
- ^ A b C d Kumar, Pankaj; Radhakrishnan, Jolly; Chowdhary, Maksud A .; Giampietro, Philip F. (01.08.2001). „Prevalence a vzorce prezentace genetických poruch na dětském pohotovostním oddělení“. Mayo Clinic Proceedings. 76 (8): 777–783. doi:10.4065/76.8.777. ISSN 0025-6196.
- ^ A b Jackson, Maria; Marks, Leah; May, Gerhard H.W .; Wilson, Joanna B. (03.12.2018). „Genetický základ nemoci“. Eseje v biochemii. 62 (5): 643–723. doi:10.1042 / EBC20170053. ISSN 0071-1365. PMC 6279436. PMID 30509934.
(počítáno z „1 ze 17“ vzácných poruch a „80%“ vzácných poruch genetických)
- ^ "Genetika rakoviny". www.medschool.lsuhsc.edu. Citováno 2020-01-14.
- ^ „OMIM Entry # 144010 - HYPERCHOLESTEROLEMIA, FAMILIAL, 2; FCHL2“. www.omim.org. Citováno 2019-07-01.
- ^ Simons, M .; Walz, G. (září 2006). „Polycystické onemocnění ledvin: Buněčné dělení bez c (l) ue?“. Ledviny International. 70 (5): 854–864. doi:10.1038 / sj.ki.5001534.
- ^ „OMIM Entry # 162200 - NEUROFIBROMATOSIS, TYP I; NF1“. www.omim.org. Citováno 2019-07-01.
- ^ Keane MG; Pyeritz RE (květen 2008). „Lékařská léčba Marfanova syndromu“. Oběh. 117 (21): 2802–13. doi:10.1161 / CIRCULATIONAHA.107.693523. PMID 18506019.
- ^ Walker FO (2007). „Huntingtonova choroba“. Lanceta. 369 (9557): 218–28 [221]. doi:10.1016 / S0140-6736 (07) 60111-1. PMID 17240289.
- ^ „OMIM Entry # 603903 - SICKLE CELL ANEMIA“. www.omim.org. Citováno 2019-07-01.
- ^ Williams T. N .; Obaro S. K. (2011). „Srpkovitá nemoc a morbidita na malárii: příběh se dvěma ocasy“. Trendy v parazitologii. 27 (7): 315–320. doi:10.1016 / j.pt.2011.02.004. PMID 21429801.
- ^ Kuliev, Anver; Verlinsky, Yury (2005). „Preimplantační diagnostika: Realistická možnost asistované reprodukce a genetické praxe“. Curr. Opin. Obstet. Gynecol. 17 (2): 179–83. doi:10.1097 / 01.gco.0000162189.76349.c5. PMID 15758612.
- ^ Simčíková D, Heneberg P (prosinec 2019). „Zpřesnění předpovědí evoluční medicíny na základě klinických důkazů o projevech Mendelianových chorob“. Vědecké zprávy. 9 (1): 18577. doi:10.1038 / s41598-019-54976-4. PMC 6901466. PMID 31819097.CS1 maint: používá parametr autoři (odkaz)
- ^ A b Griffiths, Anthony J.F .; Wessler, Susan R .; Carroll, Sean B .; Doebley, John (2012). „2: Single-Gene Inheritance“. Úvod do genetické analýzy (10. vydání). New York: W.H. Freeman a společnost. ISBN 978-1-4292-2943-2.
- ^ „Dědičné vzorce pro poruchy jednoho genu“. learn.genetics.utah.edu. Citováno 2019-07-01.
- ^ Wade, Nicholas (29. ledna 2006). "Japonští vědci identifikují gen pro ušní vosk". New York Times.
- ^ Yoshiura K; Kinoshita A; Ishida T; et al. (Březen 2006). „SNP v genu ABCC11 je určující pro typ lidského ušního mazu“. Nat. Genet. 38 (3): 324–30. doi:10.1038 / ng1733. PMID 16444273.
- ^ Mitton, Jeffery B (2002). "Heterozygotní výhoda". eLS. doi:10.1038 / npg.els.0001760. ISBN 0470016175.
- ^ Poolman EM, Galvani AP (únor 2007). „Hodnocení kandidátů na selektivní tlak na cystickou fibrózu“. Journal of the Royal Society, Interface. 4 (12): 91–8. doi:10.1098 / rsif.2006.0154. PMC 2358959. PMID 17015291.
- ^ Allison AC (říjen 2009). "Genetická kontrola odolnosti proti lidské malárii". Aktuální názor na imunologii. 21 (5): 499–505. doi:10.1016 / j.coi.2009.04.001. PMID 19442502.
- ^ Woolf, LI (1986). „Výhoda heterozygotů ve fenylketonurii“. American Journal of Human Genetics. 38 (5): 773–5. PMC 1684820. PMID 3717163.
- ^ Weatherall, D. J. (2015). „Thalassemias: Disorders of Globin Synthesis“. Williamsova hematologie (9e ed.). McGraw Hill Professional. p. 725. ISBN 9780071833011.
- ^ Nussbaum, Robert; McInnes, Roderick; Willard, Huntington (2007). Thompson & Thompson Genetics in Medicine. Philadelphia PA: Saunders. 144, 145, 146. ISBN 9781416030805.
- ^ Milunsky, Aubrey, ed. (2004). Genetické poruchy a plod: diagnostika, prevence a léčba (5. vydání). Baltimore: Johns Hopkins University Press. ISBN 978-0801879289.
- ^ „Diagnostické testy - amniocentéza“. Harvardská lékařská škola. Archivovány od originál dne 2008-05-16. Citováno 2008-07-15.
- ^ Ginn, Samantha L .; Alexander, Ian E .; Edelstein, Michael L .; Abedi, Mohammad R .; Wixon, Jo (únor 2013). „Klinické studie genové terapie po celém světě do roku 2012 - aktualizace“. The Journal of Gene Medicine. 15 (2): 65–77. doi:10,1002 / jgm.2698. PMID 23355455.
- ^ Verma, I. M. (22. srpna 2013). "Génová terapie, která funguje". Věda. 341 (6148): 853–855. Bibcode:2013Sci ... 341..853V. doi:10.1126 / science.1242551. PMID 23970689.
- ^ „Pravděpodobný genetický původ pro důlkovou hypoplázii skloviny na stoličkách Paranthropus robustus“. ResearchGate. Citováno 2019-03-09.
externí odkazy
| Klasifikace |
|---|
- Genomika veřejného zdraví ve společnosti CDC
- OMIM - Online Mendelian Inheritance in Man, katalog lidských genů a genetických poruch
- Informační centrum o genetických a vzácných onemocněních (GARD) Office of Rare Diseases (ORD), National Institutes of Health (NIH)
- Národní centrum CDC pro vrozené vady a vývojová postižení
- Informace o genetických onemocněních z projektu lidského genomu
- Globální projekt genů, organizace pro genetické a vzácné nemoci
- Seznam genetických poruch - Genome.gov