Hirschsprungova choroba - Hirschsprungs disease - Wikipedia
| Hirschsprungova choroba | |
|---|---|
| Ostatní jména | Aganglionový megakolon, vrozený megakolon, vrozená intestinální aganglionóza[1] |
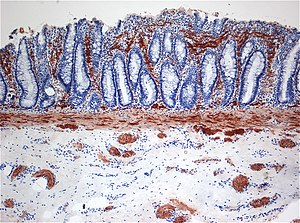 | |
| Histopatologie Hirschsprungovy choroby vykazující abnormální hodnoty acetylcholinesteráza (AchE) -pozitivní nervová vlákna (hnědá) v sliznice | |
| Specialita | Lékařská genetika |
| Příznaky | Zácpa, zvracení, bolest břicha, průjem, pomalý růst[1] |
| Komplikace | Enterokolitida, megakolon, obstrukce střev, střevní perforace[1][2] |
| Obvyklý nástup | První 2 měsíce života[1] |
| Typy | Krátký segment, dlouhý segment[1] |
| Příčiny | Genetický[1] |
| Rizikové faktory | Rodinná historie[1] |
| Diagnostická metoda | Na základě příznaků biopsie[3] |
| Diferenciální diagnostika | Chronická střevní pseudoobstrukce, mekonium ileus[2] |
| Léčba | Chirurgická operace[2] |
| Frekvence | 1 z 5 000 novorozenců[1] |
Hirschsprungova choroba (HD nebo HSCR) je vrozená vada ve kterém nervy chybí v částech střevo.[1][3] Nejvýraznějším příznakem je zácpa.[1] Jiné příznaky mohou zahrnovat zvracení, bolest břicha, průjem a pomalý růst.[1] Příznaky se obvykle projeví v prvních dvou měsících života.[1] Komplikace mohou zahrnovat enterokolitida, megakolon, obstrukce střev a střevní perforace.[1][2]
Porucha může nastat sama nebo ve spojení s jinými genetické poruchy jako Downův syndrom nebo Waardenburgův syndrom.[1][2] Asi polovina ojedinělých případů je spojena se specifickým genetickým materiálem mutace a asi 20% se vyskytuje v rodinách.[1] Některé z nich se vyskytují v autosomálně dominantní způsob.[1] Příčina zbývajících případů je nejasná.[1] Pokud jinak mají normální rodiče jedno dítě s tímto onemocněním, další dítě má 4% riziko, že bude postiženo.[2] Stav je rozdělen na dva hlavní typy, krátký segment a dlouhý segment, v závislosti na tom, kolik střeva je ovlivněno.[1] Zřídka tenké střevo mohou být také ovlivněny.[2] Diagnóza je založena na příznacích a potvrzena biopsie.[3]
Léčba se obvykle provádí chirurgickým zákrokem k odstranění postižené části střeva.[2] Nejčastěji prováděný chirurgický zákrok je znám jako „protahování“.[3] Občas, an transplantace střev může být doporučeno.[2] Hirschsprungova choroba se vyskytuje asi u jednoho z 5 000 novorozenců.[1] Muži jsou postiženi častěji než ženy.[1] Předpokládá se, že tento stav byl poprvé popsán v roce 1691 nizozemským anatomem Frederik Ruysch[4] a je pojmenována po dánském lékaři Harald Hirschsprung podle jeho popisu v roce 1888.[5][6]
Příznaky a symptomy
Typicky je Hirschsprungova choroba diagnostikována krátce po narození, i když se může vyvinout až do dospělosti kvůli přítomnosti megakolon, nebo proto, že dítě nedokáže projít první stolicí (mekonium )[7] do 48 hodin od dodání. Normálně 90% dětí projde prvním mekoniem do 24 hodin a 99% dětí do 48 hodin.[8] Mezi další příznaky patří zelené nebo hnědé zvracení, výbušné stolice poté, co lékař vloží prst do konečníku, otok břicha, nadměrné množství plynů a krvavý průjem.[Citace je zapotřebí ]
Některé případy jsou diagnostikovány později, do dětství, obvykle však před dosažením věku 10 let.[7] U dítěte se může objevit retence stolice, zácpa nebo roztažení břicha.[7]
Přidružené syndromy
Hirschsprungova choroba se také může projevit jako součást vícesystémových poruch, jako jsou:[9]
- Bardet – Biedlův syndrom
- Hypoplázie chrupavky a vlasů[10]
- Vrozený syndrom centrální hypoventilace[11]
- MUŽI2[12]
- Mowat-Wilsonův syndrom[13]
- Smith – Lemli – Opitzův syndrom[14]
- Trizomie 21 (Downův syndrom ) [15]
- Některé formy Waardenburgův syndrom
Způsobit
Porucha může nastat sama nebo ve spojení s jinými genetické poruchy jako Downův syndrom.[2] Asi polovina ojedinělých případů je spojena se specifickým genetickým materiálem mutace a asi 20% se vyskytuje v rodinách.[1] Některé z nich se vyskytují v autosomálně dominantní způsob.[1] Příčina zbývajících případů je nejasná.[1] Pokud jinak mají normální rodiče jedno dítě s tímto onemocněním, další dítě má 4% riziko, že bude postiženo.[2]
Genetika
| Typ | OMIM | Gen | Místo |
|---|---|---|---|
| HSCR1 | 142623 | RET | 10q11.2 |
| HSCR2 | 600155 | EDNRB | 13q22 |
| HSCR3 | 600837 | GDNF | 5p13.1-p12 |
| HSCR4 | 131242 | EDN3 | 20q13.2-q13.3 |
| HSCR5 | 600156 | ? | 21q22 |
| HSCR6 | 606874 | ? | 3p21 |
| HSCR7 | 606875 | ? | 19q12 |
| HSCR8 | 608462 | ? | 16q23 |
| HSCR9 | 611644 | ? | 4q31-32 |
| — | 602229 | SOX10 | 22q13 |
| — | 600423 | ECE1 | 1p36.1 |
| — | 602018 | NRTN | 19p13.3 |
| — | 602595 | GEMIN2 (Proteiny spojené s drahokamy 2 ) | 14q13-q21 |
| — | 191315 | NTRK1 | 1q23.1 |
| — | 605802 | ZEB2 | 2q22.3 |
Několik geny a specifické oblasti na chromozomech (loci ) bylo prokázáno nebo navrhováno, že je spojeno s Hirschsprungovou chorobou:
The RET protoonkogen představuje nejvyšší podíl familiárních i sporadických případů se širokou škálou mutací roztroušených po celé jeho kódující oblasti.[16] A protoonkogen může způsobit rakovinu, pokud je mutovaná nebo nadměrně exprimovaná.[17]
RET protoonkogen
RET je gen, který kóduje proteiny, které pomáhají buňky neurální lišty v jejich pohybu trávicím traktem během vývoje embrya. Tyto buňky neurální lišty nakonec tvoří svazky nervových buněk zvané gangliony. EDNRB kóduje proteiny, které spojují tyto nervové buňky s trávicím traktem. Mutace v těchto dvou genech tedy mohly přímo vést k nepřítomnosti určitých nervových vláken v tlustém střevě. Výzkum naznačuje, že s Hirschsprungovou chorobou je spojeno několik genů.[18] Nový výzkum také naznačuje, že mutace v genomových sekvencích se podílejí na regulaci EDNRB mít větší dopad na Hirschsprungovu chorobu, než se dříve myslelo.[Citace je zapotřebí ]
RET může mutovat mnoha způsoby a je spojen s Downovým syndromem. Protože Downov syndrom je komorbidní ve 2% Hirschsprungových případů, existuje pravděpodobnost, že RET se významně podílí jak na Hirschsprungově chorobě, tak na Downově syndromu. RET je také spojován s medulární rakovina štítné žlázy a neuroblastom, což je typ rakoviny běžný u dětí. Obě tyto poruchy jsou častější u Hirschsprungových pacientů než v běžné populaci. Jedna funkce REOvládací prvky T jsou pojezdy buňky neurální lišty skrz střeva ve vývoji plod. Čím dříve RET mutace se vyskytuje u Hirschsprungovy choroby, tím závažnější je porucha.
Jiné geny
Běžné a vzácné variace DNA v neuregulinu 1 (NRG1) a NRG3 (NRG3) bylo poprvé prokázáno, že je spojeno s onemocněním u čínských pacientů prostřednictvím studie asociace Genome Wide Association provedené hongkonským týmem v roce 2009 [19] respektive 2012[20] Následné studie u asijských i bělošských pacientů potvrdily počáteční nálezy hongkonské univerzity. Vzácné i běžné varianty v těchto dvou genech byly identifikovány v dalších čínštině,[21] Thajské, korejské, indonéské a španělské pacienty. Je známo, že tyto dva geny hrají roli při tvorbě enterického nervového systému; je tedy pravděpodobné, že se alespoň v některých případech budou podílet na patologii Hirschsprungovy choroby.[Citace je zapotřebí ]
Dalším genem spojeným s tímto stavem je NADPH oxidáza, EF-ruka vázající doménu vápníku 5 (NOX5 ).[22] Tento gen se nachází na dlouhém rameni chromozom 15 (15q23).
Patofyziologie
Během normálu prenatální vývoj, buňky z neurální lišty migrují do tlustého střeva (tlustého střeva) a tvoří nervové sítě zvané myenterický plexus (Auerbach plexus) (mezi hladký sval vrstvy stěny gastrointestinálního traktu) a submukózní plexus (Meissner plexus) (v submukóze stěny gastrointestinálního traktu). U Hirschsprungovy choroby není migrace úplná a část tlustého střeva je postrádá nervová těla které regulují aktivitu tlustého střeva. Dotčený segment dvojtečka nemůže relaxovat a projít stolice přes tlusté střevo, což vytváří překážku.[23]
Nejuznávanější teorií příčiny Hirschsprung je defekt v kraniokaudální migraci neuroblastů pocházející z neurální lišty, ke kterému dochází během prvních 12 týdnů těhotenství. Vady diferenciace neuroblasty k poruše může také přispívat do gangliových buněk a zrychlená destrukce gangliových buněk ve střevě.[24]
Tento nedostatek gangliových buněk v myenterickém a submukózním plexu je u Hirschsprungovy choroby dobře zdokumentován.[7] S Hirschsprungovou chorobou se segment bez neuronů (aganglionový) zúží, což způsobí, že se normální proximální část střeva roztáhne výkaly. Předpokládá se, že toto zúžení distálního tračníku a selhání relaxace v aganglionovém segmentu je způsobeno nedostatkem neuronů obsahujících syntázu oxidu dusnatého.[7]
Nejcitovanějším rysem je absence gangliových buněk: zejména u mužů nemá 75% žádné na konci tlustého střeva (rektosigmoid) a 8% nemá gangliové buňky v celém tlustém střevě. Zvětšená část střevo se nachází proximálně, zatímco zúžená, aganglionická část se nachází distálně, blíže ke konci střeva. Absence gangliových buněk vede k přetrvávající nadměrné stimulaci nervů v postižené oblasti, což vede ke kontrakci.
Ekvivalentní onemocnění u koní je syndrom smrtící bílé.[25]
Diagnóza

Definitivní diagnóza je stanovena sací biopsií distálně zúženého segmentu.[26] Histologické vyšetření tkáně by ukázalo nedostatek gangliových nervových buněk. Diagnostické techniky zahrnují anorektální manometrie,[27] baryum klystýr a rektální biopsie.Sací rektální biopsie je považována za aktuální mezinárodní Zlatý standard v diagnostice Hirschsprungovy choroby.[28]
Radiologické nálezy mohou také pomoci s diagnostikou.[29] Kinematografie (fluoroskopie z kontrastní médium procházející anorektální oblast) pomáhá při určování úrovně postižených střev.
Léčba
Léčba Hirschsprungovy choroby spočívá v chirurgickém odstranění (resekci) abnormální části tlustého střeva, po které následuje reanastomóza.[Citace je zapotřebí ]
Kolostomie
První fáze léčby bývala reverzibilní kolostomie. V tomto přístupu je zdravý konec tlustého střeva rozřezán a připojen k otvoru vytvořenému na přední straně břicha. Obsah střeva se odvádí otvorem v břiše a do vaku. Později, když jsou hmotnost, věk a stav pacienta správné, je „nový“ funkční konec střeva spojen s konečníkem. První chirurgická léčba zahrnující chirurgickou resekci následovanou reanastomózou bez kolostomie nastala již v roce 1933 doktorem Bairdem v Birmingham na ročního chlapce.[Citace je zapotřebí ]
Další postupy
Švédsko-americký chirurg, Orvar Swenson (1909–2012), který objevil příčinu Hirschsprung's, nejprve provedl chirurgickou léčbu protahovací chirurgie v roce 1948.[30] Procedura protahování opravuje tlusté střevo spojením funkční části střeva s konečníkem. Průchozí procedura je typickou metodou léčby Hirschsprung's u mladších pacientů. Swenson vymyslel původní postup a chirurgický zákrok byl mnohokrát upraven.[31]'
V současné době se používá několik různých chirurgických přístupů, mezi něž patří postupy Swenson, Soave, Duhamel a Boley.[31] Swensonova procedura ponechává malou část nemocného střeva. Soaveova procedura, pojmenovaná po italském dětském chirurgovi, Franco Soave (1917–1984), ponechává vnější stěnu tlustého střeva nezměněnou. Boleyova procedura, kterou propagoval americký chirurg, Scott Boley (b. 1941), je malá modifikace Soaveova postupu, takže se někdy používá termín „Soave-Boleyův“ postup.[32][33] Duhamelův postup, pojmenovaný pro francouzského dětského chirurga, Bernard Duhamel (1917–1996), používá a chirurgický stapler propojit dobré a špatné střevo.
U 15% dětí, které nedosáhnou úplné kontroly střev, jsou k dispozici další způsoby léčby. Zácpu lze napravit projímadly nebo dietou s vysokým obsahem vlákniny. U těchto pacientů může vážná dehydratace hrát hlavní faktor v jejich životním stylu. Nedostatek kontroly střev může být vyřešen pomocí ileostomie - podobný kolostomii, ale používá spíše konec tenkého střeva než tlustého střeva. Alternativou je také Maloneův antegrádní klystýr tlustého střeva (ACE).[34] V Malone ACE trubice prochází břišní stěnou do slepého střeva, nebo je-li k dispozici, do tlustého střeva. Potom se denně vypláchne střevo.[35] Děti ve věku 6 let si mohou tento denní nával podávat samy.
Pokud je postižená část dolního střeva omezena na dolní část konečníku, mohou být provedeny další chirurgické zákroky, například zadní rektální myektomie. Prognóza je v 70% případů dobrá. Chronická pooperační zácpa je přítomna v 7 až 8% operovaných případů. Pooperační enterokolitida, závažný projev, je přítomen u 10–20% operovaných pacientů.[Citace je zapotřebí ]
Epidemiologie
Podle studie z roku 1984 provedené v Maryland, Hirschsprungova choroba se objevuje u 18,6 na 100 000 živě narozených.[36] V Japonsku se vyskytuje podobným tempem asi u jednoho z 5 000 porodů (20 na 100 000).[37] Je častější u mužů než u žen (4,32: 1) a spíše u bílé než u bílé.[38] Devět procent případů Hirschsprung bylo také diagnostikováno s Downovým syndromem.[36] Většina případů je diagnostikována dříve, než je pacientovi 10 let.[7]
Dějiny
První zpráva o Hirschsprungově nemoci pochází z roku 1691,[39] když to popsal holandský anatom Frederik Ruysch.[40] Tato nemoc je však pojmenována po Harald Hirschsprung, dánština lékař, který poprvé popsal dvě kojence, kteří zemřeli na tuto poruchu v roce 1888.[5][6]
Hirschsprungova choroba je kongenitální porucha tlustého střeva, při které jsou některé nervové buňky známé jako gangliové buňky, chybí, způsobují chronický zácpa.[41] U pacientů s Hirschsprungovou chorobou chybí jak myenterické, tak submukózní plexy.[42] Barium klystýr je základním pilířem diagnostiky Hirschsprung, ačkoli rektální biopsie ukazující nedostatek gangliových buněk je jedinou jistou metodou diagnostiky.[Citace je zapotřebí ]
První publikace o důležitém genetickém objevu choroby byla od Martucciello Giuseppe et al. v roce 1992. Autoři popsali případ pacienta s celkovou aganglionózou tlustého střeva spojenou s karyotypem 46, XX, del 10 (q11,21 q21,2).[43] V této chromozomální oblasti 10 byl identifikován hlavní gen Hirschsprungovy choroby, byl to protoonkogen RET.[44]
Obvyklou léčbou je chirurgický zákrok, při kterém se část tlustého střeva, která má nervové buňky, protáhne a přišije přes část, která postrádá nervové buňky.[45] Po dlouhou dobu byl Hirschsprung’s považován za multifaktoriální poruchu, kde byla za příčinu považována kombinace přírody a výchovy. V srpnu 1993 však byly v nezávislých skupinách zveřejněny dva články Genetika přírody řekl, že Hirschsprungovu chorobu lze zmapovat do určité míry chromozom 10.[46][47]
Tento výzkum také naznačil, že za poruchu byl zodpovědný jediný gen. Vědci to však nedokázali izolovat.
Viz také
- Achalasie
- Ileus, selhání peristaltické svalové aktivity ve střevě
- Střevní neuronální dysplázie
Reference
- ^ A b C d E F G h i j k l m n Ó p q r s t u proti w „Hirschsprungova choroba“. Genetická domácí reference. Srpna 2012. Citováno 14. prosince 2017.
- ^ A b C d E F G h i j k „Hirschsprungova choroba“. Informační centrum o genetických a vzácných onemocněních (GARD) - program NCATS. 2017. Citováno 14. prosince 2017.
- ^ A b C d „Hirschsprungova choroba“. NORD (Národní organizace pro vzácné poruchy). 2017. Citováno 14. prosince 2017.
- ^ Holschneider, Alexander Matthias; Puri, Prem (2007). Hirschsprungova choroba a spojenecké poruchy. Springer Science & Business Media. str. 1. ISBN 9783540339359.
- ^ A b „Hirschsprungova choroba“. www.whonamedit.com. Citováno 8. října 2019.
- ^ A b Hirschsprung, H. (1888). „Stuhlträgheit Neugeborener in Folge von Dilatation und Hypertrophie des Colons“. Jahrbuch für Kinderheilkunde und physische Erziehung. Berlín. 27: 1–7.
- ^ A b C d E F Goldman, Lee (2012). Goldmanova cecilová medicína (24. vydání). Philadelphia: Elsevier Saunders. str. 867. ISBN 978-1437727883.
- ^ Kimura, Ken; Loening-Baucke, Vera (01.11.1999). „Selhání při průchodu mekoniem: Diagnostika neonatální střevní obstrukce“. Americký rodinný lékař. 60 (7): 2043–50. ISSN 0002-838X. PMID 10569507.
- ^ Online Mendelian Inheritance in Man (OMIM): 142623
- ^ Mäkitie O, Heikkinen M, Kaitila I, Rintala R (2002). „Hirschsprungova choroba v hypoplázii chrupavky a vlasů má špatnou prognózu“. J Pediatr Surg. 37 (11): 1585–8. doi:10.1053 / jpsu.2002.36189. PMID 12407544.
- ^ de Pontual L, Pelet A, Clement-Ziza M, Trochet D, Antonarakis SE, Attie-Bitach T, Beales PL, Blouin JL, Dastot-Le Moal F, Dollfus H, Goossens M, Katsanis N, Touraine R, Feingold J, Munnich A, Lyonnet S, Amiel J (2007). "Epistatické interakce s běžnou hypomorfní alelou RET u syndromické Hirschsprungovy choroby". Lidská mutace. 28 (8): 790–6. doi:10.1002 / humu.20517. PMID 17397038.
- ^ Saunders CJ, Zhao W, Ardinger HH (2009). „Komplexní analýza genů ZEB2 pro Mowat-Wilsonův syndrom v severoamerické kohortě: navrhovaný přístup k molekulární diagnostice“. American Journal of Medical Genetics. 149A (11): 2527–31. doi:10,1002 / ajmg.a.33067. PMID 19842203. S2CID 22472646.
- ^ Bonnard A, Zeidan S, Degas V, Viala J, Baumann C, Berrebi D, Perrusson O, El Ghoneimi A (2009). "Výsledky Hirschsprungovy choroby spojené s Mowat-Wilsonovým syndromem". Journal of Pediatric Surgery. 44 (3): 587–91. doi:10.1016 / j.jpedsurg.2008.10.066. PMID 19302864.
- ^ Mueller C, Patel S, Irons M, Antshel K, Salen G, Tint GS, Bay C (2003). „Normální poznání a chování u pacienta se syndromem Smith-Lemli-Opitz, u kterého se projevila Hirschsprungova choroba“. American Journal of Medical Genetics. 123A (1): 100–6. doi:10,1002 / ajmg.a.20491. PMC 1201564. PMID 14556255.
- ^ Flori E, Girodon E, Samama B, Becmeur F, Viville B, Girard-Lemaire F, Doray B, Schluth C, Marcellin L, Boehm N, Goossens M, Pingault V (2005). „Mozaicismus trizomie 7, mateřská uniparentální heterodisomie 7 a Hirschsprungova choroba u dítěte se Silver-Russellovým syndromem“. European Journal of Human Genetics. 13 (9): 1013–8. doi:10.1038 / sj.ejhg.5201442. PMID 15915162.
- ^ Martucciello G, Ceccherini I, Lerone M, Jasonni V (2000). „Patogeneze Hirschsprungovy choroby“. Journal of Pediatric Surgery. 35 (7): 1017–1025. doi:10.1053 / jpsu.2000.7763. PMID 10917288.
- ^ Chial, H (2008). „Protoonkogeny k onkogenům k rakovině“. Přírodní výchova. 1: 33.
- ^ Puri P, Shinkai T (2004). „Patogeneze Hirschsprungovy choroby a její varianty: nedávný pokrok“. Semin. Pediatr. Surg. 13 (1): 18–24. doi:10.1053 / j.sempedsurg.2003.09.004. PMID 14765367. S2CID 11395791.
- ^ Garcia-Barcelo, Maria-Merce (2009). „Celomanomová asociační studie identifikuje NRG1 jako lokus citlivosti na Hirschsprungovu chorobu“. Proc. Natl. Acad. Sci. USA. 106 (8): 2694–2699. Bibcode:2009PNAS..106,2694G. doi:10.1073 / pnas.0809630105. PMC 2650328. PMID 19196962.
- ^ Tang, Clara (10. května 2012). „Analýza počtu kopií v genomu odhaluje nový gen HSCR: NRG3“. PLOS Genet. 8 (5): e1002687. doi:10.1371 / journal.pgen.1002687. PMC 3349728. PMID 22589734.
- ^ Yang J, Duan S, Zhong R, Yin J, Pu J, Ke J, Lu X, Zou L, Zhang H, Zhu Z, Wang D, Xiao H, Guo A, Xia J, Miao X, Tang S, Wang G (2013). „Exome sekvenování identifikovalo NRG3 jako nový citlivý gen Hirschsprungovy choroby v čínské populaci“. Mol. Neurobiol. 47 (3): 957–66. doi:10.1007 / s12035-012-8392-4. PMID 23315268. S2CID 16842806.
- ^ Shin JG, Seo JY, Seo JM, Kim DY, Oh JT, Park KW, Kim HY, Kim JH, Shin HD (2019) Asociační analýza polymorfismů NOX5 s Hirschsprungovou chorobou. J Pediatr Surg
- ^ Parisi MA, Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (2002). Pagon RA, Bird TC, Dolan CR, Stephens K. (eds.). "Hirschsprung Disease Overview". GeneReviews. PMID 20301612.
- ^ Kays DW (1996). „Chirurgické stavy střevního traktu novorozence“. Kliniky v perinatologii. 23 (2): 353–75. doi:10.1016 / S0095-5108 (18) 30246-X. PMID 8780909.
- ^ Metallinos DL, Bowling AT, Rine J (červen 1998). „Missense mutace v genu pro endotelinový B receptor je spojena s Lethal White Foal Syndrome: koňská verze Hirschsprungovy choroby“. Mamm. Genom. 9 (6): 426–31. doi:10,1007 / s003359900790. PMID 9585428. S2CID 19536624. Archivovány od originál dne 2000-09-16.
- ^ Dobbins WO, Bill AH (1965). „Diagnóza Hirschsprungovy choroby vyloučené biopsií rektálního sání“. New England Journal of Medicine. 272 (19): 990–993. doi:10.1056 / NEJM196505132721903. PMID 14279253.
- ^ Eli Ehrenpreis (říjen 2003). Vysvětlení análních a rektálních onemocnění. Remedica. str. 15–. ISBN 978-1-901346-67-1. Citováno 2010-11-12.
- ^ Martucciello G, Pini Prato A, Puri P, Holschneider AM, Meier-Ruge W, Jasonni V, Tovar JA, Grosfeld JL (2005). „Spory týkající se diagnostických pokynů pro anomálie střevního nervového systému: zpráva ze čtvrtého mezinárodního sympozia o Hirschsprungově nemoci a souvisejících neurocristopathiích“. J Pediatr Surg. 40 (10): 1527–31. doi:10.1016 / j.jpedsurg.2005.07.053. PMID 16226977.
- ^ Kim HJ, Kim AY, Lee CW, Yu CS, Kim JS, Kim PN, Lee MG, Ha HK (2008). „Hirschsprungova choroba a hypoganglionóza u dospělých: radiologické nálezy a diferenciace“. Radiologie. 247 (2): 428–34. doi:10,1148 / radiol.2472070182. PMID 18430875.
- ^ Swenson O (1989). „Moje rané zkušenosti s Hirschsprungovou chorobou“. J. Pediatr. Surg. 24 (8): 839–44, diskuse 844–5. doi:10.1016 / S0022-3468 (89) 80549-4. PMID 2671336.
- ^ A b „Hirschsprungova choroba“. Americká pediatrická chirurgická asociace. Citováno 11. června 2019.
- ^ W. Allan Walker (01.07.2004). Pediatrické gastrointestinální onemocnění: patofyziologie, diagnostika, léčba. PMPH-USA. 2120–. ISBN 978-1-55009-240-0. Citováno 2010-11-12.
- ^ Timothy R. Koch (2003). Nemoci tlustého střeva. Humana Press. 387–. ISBN 978-0-89603-961-2. Citováno 2010-11-12.
- ^ Malone PS, Ransley PG, Kiely EM (1990). "Předběžná zpráva: antegrádní klystýr". Lanceta. 336 (8725): 1217–1218. doi:10.1016/0140-6736(90)92834-5. PMID 1978072. S2CID 9203632.
- ^ Walsh RA, Koyle MA, Waxman SW (2000). „Malone ACE postup pro fekální inkontinenci“. Infekce v urologii. 13 (4).
- ^ A b Goldberg EL (1984). „Epidemiologická studie Hirschsprungovy choroby“. Int J Epidemiol. 13 (4): 479–85. doi:10.1093 / ije / 13.4.479. PMID 6240474.
- ^ Suita S, Taguchi T, Ieiri S, Nakatsuji T (2005). „Hirschsprungova choroba v Japonsku: analýza 3852 pacientů na základě celostátního průzkumu za 30 let“. Journal of Pediatric Surgery. 40 (1): 197–201, diskuse 201–2. doi:10.1016 / j.jpedsurg.2004.09.052. PMID 15868585.
- ^ Colwell, Janice (2004). Správa fekální a močové přesměrování. Mosby. str. 264. ISBN 978-0-323-02248-4.
- ^ Hirschsprungova choroba a spojenecké poruchy. Berlín: Springer. 2007. ISBN 978-3-540-33934-2.
- ^ „Hirschsprungova choroba: pozadí, patofyziologie, epidemiologie“. 2017-01-08. Citovat deník vyžaduje
| deník =(Pomoc) - ^ Worman S, Ganiats TG (1995). „Hirschsprungova choroba: příčina chronické zácpy u dětí“. Jsem známý lékař. 51 (2): 487–94. PMID 7840044.
- ^ https://www.medscape.com/answers/178493-62797/what-is-the-pathophysiology-of-hirschsprung-disease#qna
- ^ Martucciello G; Bicocchi MP; Dodero P .; Lerone M .; Silengo Cirillo M; Puliti A; Gimelli G; Romeo G. (1992). „Celková aganglionóza tlustého střeva spojená s intersticiální delecí dlouhého ramene chromozomu 10“. Pediatrická chirurgie International. 7 (4): 308–310. doi:10.1007 / BF00183991. S2CID 40123658.
- ^ Romeo G, Ronchetto P, Luo Y, Barone V, Seri M, Ceccherini I, Pasini B, Bocciardi R, Lerone M, Kääriäinen H (1994). „Bodové mutace ovlivňující tyrosinkinázovou doménu RET protoonkogenu u Hirschsprungovy choroby“. Příroda. 367 (6461): 377–378. Bibcode:1994Natur.367..377R. doi:10.1038 / 367377a0. PMID 8114938. S2CID 157274.
- ^ (Národní informační středisko zažívacích chorob).
- ^ Angrist M, Kauffman E, Slaugenhaupt SA, Matise TC, Puffenberger EG, Washington SS, Lipson A, Cass DT, Reyna T, Weeks DE (1993). „Gen pro Hirschsprungovu chorobu (megakolon) v pericentromerické oblasti lidského chromozomu 10“. Nat. Genet. 4 (4): 351–6. doi:10.1038 / ng0893-351. PMID 8401581. S2CID 22031571.
- ^ Lyonnet S, Bolino A, Pelet A, Abel L, Nihoul-Fékété C, Briard ML, Mok-Siu V, Kaariainen H, Martucciello G, Lerone M, Puliti A, Luo Y, Weissenbach J, Devoto M, Munnich A, Romeo G (1993). „Gen pro Hirschsprungovu chorobu se mapuje do proximálního dlouhého ramene chromozomu 10“. Nat. Genet. 4 (4): 346–50. doi:10.1038 / ng0893-346. PMID 8401580. S2CID 29089707.
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |