Robinowův syndrom - Robinow syndrome
| Robinowův syndrom | |
|---|---|
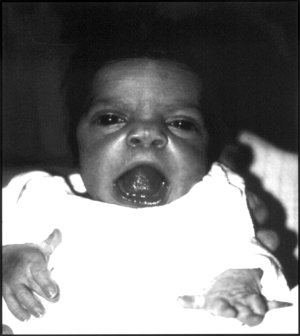 | |
| Dítě vykazující rysy obličeje Robinowova syndromu. | |
| Specialita | Lékařská genetika |
Robinowův syndrom je extrémně vzácný genetická porucha vyznačuje se krátkými končetinami nanismus abnormality v hlavě, obličeji a vnějšku genitálie, stejně jako vertebrální segmentace. Porucha byla poprvé popsána v roce 1969 člověkem genetik Meinhard Robinow,[1] spolu s lékaři Frederic N. Silverman a Hugo D. Smith, v American Journal of Diseases of Children. Do roku 2002 bylo zdokumentováno a zavedeno do lékařské literatury více než 100 případů.[1]
Existují dvě formy poruchy, dominantní a recesivní, z nichž první je častější. Pacienti s dominantní verzí často trpí výše zmíněnými příznaky. Recesivní případy jsou na druhou stranu obvykle fyzicky výraznější a jednotlivci mohou vykazovat více kosterní abnormality.[2] Recesivní forma je obzvláště častá v krocan.[3] To však lze pravděpodobně vysvětlit a společný předek, protože rodiny těchto pacientů lze vysledovat do jediného města ve východním Turecku.[4] Klastry autosomální recesivní forma byla také dokumentována v Omán a Československo.[1]
Tento syndrom je také známý jako Robinow-Silverman-Smithův syndrom, Robinowův nanismus, obličej plodu, syndrom obličeje plodu,[5] syndrom fetální facie, akrální dysostóza s abnormalitami obličeje a genitálií nebo syndrom mezomelického nanismu - malé genitálie.[6] Recesivní forma byla dříve známá jako Covesdemův syndrom.
Příznaky a symptomy


Robinow si všiml podobnosti tváří postižených pacientů s tváří a plod používající výraz „fetální facie“ k popisu vzhledu malého obličeje a široce rozmístěných očí.[1] Klinické rysy mohou také zahrnovat krátký, vztyčený nos, výrazné čelo a plochý nosní můstek. Horní ret může být "stanovaný",[1] odhalování shlukování zubůkravata na jazyk „nebo guma hypertrofie.
Ačkoli oči nevyčnívají, abnormality v dolní části oční víčko může působit dojmem. Pokud se oči nemohou úplně zavřít, může být nutná operace. Kromě toho uši může být nízko položený na hlavě nebo deformovaný boltce.[Citace je zapotřebí ]
Pacienti trpí nanismem, krátké paže, malé nohy a malé ruce. Prsty a prsty mohou být také neobvykle krátká a laterálně nebo mediálně ohnuté. Palec může být posunutý a někteří pacienti, zejména v Turecku, mají zkušenosti ektrodaktylie.[1] Všichni pacienti často trpí abnormalitami segmentace obratlů. Ti, kteří mají dominantní variantu, mají nanejvýš jediný motýlí obratle.[2] Ti, kteří mají recesivní formu, však mohou trpět hemivertebrae, vertebrální fúze a anomálie žeber. Některé případy se podobají Jarcho-Levinův syndrom nebo spondylocostální dysostóza.[Citace je zapotřebí ]
K pohlavním vadám, které jsou typicky patrné u mužů, patří a mikropenis s normálně vyvinutým šourek a testy. Někdy mohou být varlata nesestoupená nebo může pacient trpět hypospadias.[2] Poruchy ženských pohlavních orgánů mohou zahrnovat zmenšenou velikost klitoris a málo rozvinutý stydké pysky. Zřídka stydké pysky může být také málo rozvinutý.[2] Některé výzkumy ukázaly, že ženy mohou zažít vaginální atrézie nebo hematocolpos.[3]
Autozomálně recesivní forma poruchy má tendenci být mnohem závažnější. Příklady rozdílů jsou shrnuty v následující tabulce:[7]
| Charakteristický | Autosomálně recesivní | Autozomálně dominantní |
|---|---|---|
| Postava | Kratší postava -2 SD nebo méně | Krátké nebo normální |
| Zbraně | Velmi krátké | Mírně krátké |
| Loket | Radiální dislokace hlavy | Žádná radiální dislokace hlavy |
| Horní ret | Stanovaný horní ret | Normální horní ret |
| Úmrtnost | 10% úmrtnost | Žádná nadměrná úmrtnost |
Související podmínky
Mezi zdravotní stavy patří časté ušní infekce ztráta sluchu, hypotonie, vývojové problémy, dýchací potíže, potíže s jídlem, citlivost na světlo, a reflux jícnu.[2]
Údaje o plodnost a rozvoj sekundární sexuální charakteristiky je poměrně řídký. Bylo hlášeno, že jak muži, tak ženy měli děti. Muži, kteří se rozmnožovali, měli všichni autozomálně dominantní formu poruchy; plodnost pacientů s recesivní variantou není známa.[1]
Vědci také hlásili abnormality v ledvinový trakt postižených pacientů. Hydronefróza je relativně běžný stav a vědci předpokládali, že to může vést k infekce močového ústrojí.[8] Kromě toho trpěla řada pacientů cystická dysplázie z ledviny.[1]
S Robinowovým syndromem je často spojena řada dalších stavů. Asi 15% hlášených pacientů trpí vrozené srdeční vady. Ačkoli neexistuje jasný vzor, patří mezi nejběžnější podmínky plicní stenóza a atrézie.[9] Navíc, i když je inteligence obecně normální, zhruba u 15% pacientů dochází ke zpoždění ve vývoji.[1]
Genetika
Genetický studie spojily autozomálně recesivní formu poruchy s ROR2 gen na pozici 9 dlouhého ramene chromozom 9.[1] Tento gen je zodpovědný za aspekty růstu kostí a chrupavek. Stejný gen se podílí na vzniku autozomálně dominantního brachydactyly B.[1]

Autosomálně dominantní forma byla spojena se třemi geny - WNT5A, Protein polarity segmentu rozcuchaný homolog DVL-1 (DVL1 ) a protein polarity segmentu rozcuchaný homolog DVL-3 (DVL3 ). Tato forma je často způsobena novými mutacemi a je obecně méně závažná než recesivní forma. S touto poruchou byly spojeny dva další geny - Frizzled-2 (FZD2 ) a Nucleoredoxin (Gen NXN ).[10] Všechny tyto geny patří do stejné metabolické dráhy - systému WNT. Tento systém se podílí na sekreci různých sloučenin jak u plodu, tak u dospělých.[Citace je zapotřebí ]
Fetální ultrazvuk může nabídnout prenatální diagnostika 19 týdnů do těhotenství. Charakteristiky plodu trpícího mírnější dominantní formou však nemusí být vždy snadné odlišit od závažnějšího recesivního případu. Genetické poradenství je možnost vzhledem k dostupnosti rodinné historie.[1]
Diagnóza
Robinowův syndrom je podezřelý z klinických nálezů a rodinné anamnézy a potvrzen typickými bialelickými patogenními variantami ROR-2 identifikovanými molekulárně genetickým testováním.[11]
Léčba
Léčba různých projevů bude obvykle řešena multidisciplinárním týmem.[12]
Dějiny
Porucha byla poprvé popsána v roce 1969 německo-americkým člověkem Genetik Meinhard Robinow (1909–1997),[1] spolu s lékaři Fredericem N. Silvermanem a Hugem D. Smithem v American Journal of Diseases of Children. Do roku 2002 bylo zdokumentováno a zavedeno do lékařské literatury více než 100 případů.[1]
Reference
- ^ A b C d E F G h i j k l m n Patton, MA; Afzal, A. R (2002). "Robinowův syndrom". Journal of Medical Genetics. 39 (5): 305–10. doi:10,1136 / jmg. 39.5.305. PMC 1735132. PMID 12011143.
- ^ A b C d E Nadace Robinowova syndromu. Obecná informace. Přístupné 19. května 2006.
- ^ A b Balci, Sevim; Beksaç, Sinan; Haliloglu, Mithat; Ercis, Murat; Eryilmaz, Muzaffer (1998). "Robinowův syndrom, vaginální atrézie, hematocolpos a extra prostředníček". American Journal of Medical Genetics. 79 (1): 27–9. doi:10.1002 / (SICI) 1096-8628 (19980827) 79: 1 <27 :: AID-AJMG7> 3.0.CO; 2-F. PMID 9738864.
- ^ Brunner, Han G; Van Bokhoven, Hans; Celli, Jacopo; Kayserili, Hülya; Van Beusekom, Ellen; Balci, Sevim; Brussel, Wim; Skovby, Flemming; Kerr, Bronwyn; Percin, E. Ferda; Akarsu, Nurten (2000). „Mutace genu kódujícího tyrosinkinázu ROR2 způsobuje autosomálně recesivní Robinowův syndrom“. Genetika přírody. 25 (4): 423–6. doi:10.1038/78113. PMID 10932187.
- ^ Národní organizace pro vzácné poruchy, Inc. Robinowův syndrom. Poslední úprava 15. května 2006. Přístup k 19. května 2006.
- ^ Databáze syndromů Jablonski. Syndromy vrozené anomálie / mentální retardace (MCA / MR). Zpřístupněno 20. května 2006.
- ^ Robinow, M (1993). "Robinowův syndrom (fetální obličej)". Klinická dysmorfologie. 2 (3): 189–98. doi:10.1097/00019605-199307000-00001. PMID 8287180.
- ^ Shprintzen, Robert J; Goldberg, R. B; Saenger, P; Sidoti, E. J (1982). „Přenos Robinowova syndromu z muže na muže“. American Journal of Diseases of Children. 136 (7): 594–7. doi:10.1001 / archpedi.1982.03970430026007. PMID 7091086.
- ^ Webber, Steven A; Wargowski, David S; Chitayat, David; Sandor, George G. S (1990). „Vrozená srdeční choroba a Robinowův syndrom: náhoda nebo další složka syndromu?“. American Journal of Medical Genetics. 37 (4): 519–21. doi:10,1002 / ajmg.1320370418. PMID 2260599.
- ^ White, Janson J; Mazzeu, Juliana F; Coban-Akdemir, Zeynep; Bayram, Yavuz; Bahrambeigi, Vahid; Hoischen, Alexander; Van Bon, Bregje W.M .; Gezdirici, Alper; Gulec, Elif Yilmaz; Ramond, Francis; Touraine, Renaud; Thevenon, Julien; Shinawi, Marwan; Bobr, Erin; Heeley, Jennifer; Hoover-Fong, Julie; Durmaz, Ceren D; Karabulut, Halil Gurhan; Marzioglu-Ozdemir, Ebru; Cayir, Atilla; Duz, Mehmet B; Sedm, Mehmet; Price, Susan; Ferreira, Barbara Merfortová; Vianna-Morgante, Angela M; Ellard, Sian; Parrish, Andrew; Stals, Karen; Flores-Daboub, Josue; et al. (2018). „Poruchy signalizace WNT jsou základem genetické heterogenity Robinowova syndromu“. American Journal of Human Genetics. 102 (1): 27–43. doi:10.1016 / j.ajhg.2017.10.002. PMC 5777383. PMID 29276006.
- ^ Afzal AR, Jeffery S (červenec 2003). „Jeden gen, dva fenotypy: mutace ROR2 u autozomálně recesivního Robinowova syndromu a autozomálně dominantní brachydaktylie typu B“. Hučení. Mutat. 22 (1): 1–11. doi:10.1002 / humu.10233. PMID 12815588.
- ^ Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, Roifman M, Brunner H, Lohr J, Mazzeu J, Chitayat D (říjen 2019). Autosomální dominantní Robinowův syndrom v GeneReviews. PMID 25577943.
Další čtení
- White, Janson; Mazzeu, Juliana F; Hoischen, Alexander; Jhangiani, Shalini N; Gambin, Tomasz; Alcino, Michele Calijorne; Penney, Samantha; Saraiva, Jorge M; Hove, Hanne; Skovby, Flemming; Kayserili, Hülya; Estrella, Elicia; Vulto-Van Silfhout, Anneke T; Steehouwer, Marloes; Muzny, Donna M; Sutton, V. Reid; Gibbs, Richard A; Lupski, James R; Brunner, Han G; Van Bon, Bregje W.M .; Carvalho, Claudia M.B (2015). „Shlukování mutací rámce posunu DVL1 v předposledním exonu způsobuje autosomálně dominantní Robinowův syndrom“. American Journal of Human Genetics. 96 (4): 612–22. doi:10.1016 / j.ajhg.2015.02.015. PMC 4385180. PMID 25817016.
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |
- ID nemoci 5704 na NIH kancelář Vzácné nemoci