Inkontinentia pigmenti - Incontinentia pigmenti
| Inkontinentia pigmenti | |
|---|---|
| Ostatní jména | Bloch – Siemensův syndrom, Bloch – Sulzbergerova choroba, Bloch – Sulzbergerův syndrom, nelanoblastosis cutis, nevus pigmentosus systematicus[1] |
 | |
| Tato podmínka se dědí v X-vázaný dominantní způsob. | |
| Specialita | Lékařská genetika |
Inkontinentia pigmenti (IP) je vzácný X-vázaný dominantní genetická porucha který ovlivňuje pokožku, vlasy, zuby, nehty a centrální nervový systém. Je pojmenován podle svého vzhledu pod mikroskopem.[1]
Toto onemocnění je charakterizováno kožními abnormalitami, které začínají v dětství, obvykle puchýřkovitou vyrážkou, která se hojí, následovanou vývojem tvrdších kožních výrůstků. Na pokožce se mohou objevit šedé nebo hnědé skvrny, které časem blednou. Mezi další příznaky patří ztráta vlasů, abnormality zubů, abnormality očí, které mohou vést ke ztrátě zraku a lemované nebo důlkové nehty a nehty. Související problémy mohou zahrnovat opožděný vývoj, mentální postižení, záchvaty a další neurologické problémy. Většina mužů s tímto onemocněním nepřežije do porodu.
Inkontinentia pigmenti je způsobena mutací v IKBKG gen, který kóduje NEMO protein, který slouží k ochraně buněk proti TNF-alfa -indukovaný apoptóza. Nedostatek IKBKG proto činí buňky náchylnější k apoptóze.
Neexistuje žádná specifická léčba; jednotlivé podmínky musí zvládnout odborníci.[2]
Prezentace
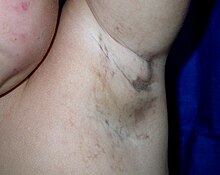
The kožní léze vyvíjejí se prostřednictvím charakteristických fází:
- puchýře (od narození do přibližně čtyř měsíců věku),
- A bradavice -jako vyrážka (několik měsíců),
- vířící makulární hyperpigmentace (přibližně od šesti měsíců do dospělosti), následovaný
- lineární hypopigmentace.
Alopecie, jsou pozorovány anomálie zubů a dystrofické nehty. Někteří pacienti mají predisponující vaskulární abnormality sítnice oddělení sítnice v raném dětství. Kognitivní zpoždění nebo mentální postižení jsou občas vidět.[Citace je zapotřebí ]
Zbarvená kůže je způsobena nadměrnými usazeninami melanin (normální kůže pigment U většiny novorozenců s IP se během prvních dvou týdnů vytvoří zabarvená kůže. Pigmentace zahrnuje kmen a končetiny, je břidlicově šedá, modrá nebo hnědá a je distribuována nepravidelně mramorovanými nebo vlnitými čarami. Odbarvení někdy s věkem mizí.[Citace je zapotřebí ]
Neurologické problémy mohou zahrnovat mozková atrofie, tvorba malých dutin v centrální bílé hmotě mozku a ztráta neurony v mozková kůra Asi 20% dětí s IP bude mít pomalé vývoj motoru svalová slabost na jedné nebo obou stranách těla, mentální postižení a záchvaty. Pravděpodobně budou mít také vizuální problémy, které mohou zahrnovat: zkříženýma očima, šedý zákal, oddělení sítnice a těžká ztráta zraku. Časté jsou také zubní problémy hypodoncie, neobvykle tvarované zuby a opožděné erupce zubu.[3]
Anomálie prsu se mohou objevit u 1% pacientů a mohou zahrnovat hypoplázie nebo nadpočetné bradavky.
Skeletální a strukturální anomálie se mohou vyskytnout přibližně u 14% pacientů, včetně:[Citace je zapotřebí ]
- Somatická asymetrie
- Hemivertebrae
- Skolióza
- Spina bifida
- Syndactyly
- Acheiria (kongenitální absence rukou - poznámka: mohou být postiženy jiné končetiny)
- Ušní anomálie
- Extra žebra
- Deformace lebky
Genetika
IP se dědí dominantním způsobem spojeným s X.[4][5] IP je u většiny mužů smrtelná. Žena s IP mohla zdědit mutaci IKBKG od kteréhokoli z rodičů nebo mít novou genovou mutaci. Rodiče mohou být klinicky postiženi nebo mohou mít zárodečnou linii mozaicismus. Postižené ženy mají 50% riziko přenosu mutované alely IKBKG při početí; většina postižených mužských konceptů však potratí. Efektivní poměr tedy u živě narozených dětí od matky nesoucí mutaci je 33% neovlivněných žen, 33% postižených žen a 33% neovlivněných mužů. Genetické poradenství, prenatální testování a preimplantační genetická diagnostika je k dispozici.[Citace je zapotřebí ]
U žen byly buňky exprimující mutovaný gen IKBKG způsobeny lyonizace selektivně umírá v době narození, takže X-inaktivace je extrémně vysoká zkosený.[6]
IP je způsobena mutacemi v genu zvaném NEMO (NF-kB základní modulátor).
Diagnóza
Diagnóza IP je stanovena na základě klinických nálezů a příležitostně na základě potvrzující biopsie kůže. Molekulárně genetické testování NEMO IKBKG Gen (chromozomální lokus Xq28) odhaluje mutace způsobující onemocnění asi u 80% probandů. Takové testování je k dispozici klinicky. Navíc ženy s IP mají zkreslenou inaktivaci X-chromozomu; k podpoře diagnózy lze použít testování. Mnoho lidí v minulosti dostalo nesprávnou diagnózu druhého typu IP, dříve známého jako IP1. Toto nyní dostalo své vlastní jméno - “Hypomelanóza Ito ' (inkontinentia pigmenti achromians ). To má trochu jinou prezentaci: víry nebo pruhy hypopigmentace a depigmentace. to je ne zděděné a nezahrnuje kožní stadia 1 nebo 2. Asi 33–50% pacientů má multisystémové postižení - oční, kosterní a neurologické abnormality. Jeho chromozomální lokus je na Xp11, spíše než Xq28.
Léčba
Specifické zacházení s IP zatím neexistuje. Léčba může řešit pouze jednotlivé příznaky.[7]
Dějiny
Tuto poruchu poprvé ohlásil švýcarský dermatolog Bruno Bloch v roce 1926 a americký dermatolog Marion Sulzberger v roce 1928.[8][9][2]
Viz také
- Seznam kožních stavů
- Seznam rentgenových nálezů spojených s kožními stavy
- Seznam zubních abnormalit spojených s kožními stavy
Reference
- ^ A b Rapini, Ronald P .; Bolognia, Jean L .; Jorizzo, Joseph L. (2007). Dermatologie: dvoudílná sada. St. Louis: Mosby. ISBN 978-1-4160-2999-1.[stránka potřebná ]
- ^ A b Sulzberger, Marion B (1928). „Über eine bisher nicht beschriebene congenitale Pigmentanomalie“ [O dříve popsané vrozené anomálii pigmentu]. Archiv für Dermatologie und Syfilis (v němčině). 154: 19–32. doi:10.1007 / bf01828398. S2CID 40446256.
- ^ Minić, S; Trpinac, D; Gabriel, H; Gencik, M; Obradović, M (leden 2013). „Zubní a orální anomálie u inkontinentních pigmentů: systematický přehled“. Clin Oral Investig. 17 (1): 1–8. doi:10.1007 / s00784-012-0721-5. PMID 22453515. S2CID 73197872.
- ^ Pettigrew, Rachel; Kuo, Hung-Chih; Scriven, Paul; Rowell, Paula; Pal, Kalyani; Handyside, Alan; Braude, Peter; Ogilvie, Caroline Mackie (2000). „Těhotenství po PGD u autosomálně dominantního inkontinentního pigmentu s invazí na X (Bloch-Sulzbergerův syndrom): kazuistika“. Lidská reprodukce. 15 (12): 2650–2. doi:10.1093 / humrep / 15.12.2650. PMID 11098039.
- ^ „Incontinentia pigmenti. DermNet NZ“.
- ^ Konsorcium International Incontinentia Pigmenti (IP); Smahi, Asmae; Courtois, G; Vabres, P; Yamaoka, S; Heuertz, S; Munnich, A; Israël, A; Heiss, Nina S; Klauck, S. M; Kioschis, P; Wiemann, S; Poustka, A; Esposito, Teresa; Bardaro, T; Gianfrancesco, F; Ciccodicola, A; d'Urso, M; Woffendin, Hayley; Jakins, T; Donnai, D; Stewart, H; Kenwrick, S. J; Aradhya, Swaroop; Yamagata, T; Levy, M; Lewis, R. A; Nelson, D. L (2000). „Genomické přeskupení v NEMO zhoršuje aktivaci NF-κB a je příčinou inkontinentní pigmenti“. Příroda. 405 (6785): 466–72. Bibcode:2000Natur.405..466T. doi:10.1038/35013114. PMID 10839543. S2CID 186243924.
- ^ „Incontinentia pigmenti“. Medline Plus. Citováno 26. prosince 2017.
- ^ Bloch-Sulzbergerova pigmentová dermatóza (Bruno Bloch) na Kdo to pojmenoval?
- ^ Bloch, B. (1926). „Eigentümliche, bisher nicht beschriebene Pigmentaffektion (incontinentia pigmenti)“ [[Zvláštní, dosud nevysvětlitelná pigmentová náklonnost (inkontinentia pigmenti)]. Schweizerische medizinische Wochenschrift (v němčině). Basilej. 56: 404–5.
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |