Mitochondriální nemoc - Mitochondrial disease
tento článek potřebuje další citace pro ověření. (únor 2014) (Zjistěte, jak a kdy odstranit tuto zprávu šablony) |
| Mitochondriální nemoc | |
|---|---|
 | |
| Mikrograf ukazující otrhaná červená vlákna, nález pozorovaný u různých typů mitochondriálních onemocnění. Svalová biopsie. Gomori trichromová skvrna. | |
| Specialita | Lékařská genetika |
Mitochondriální nemoci jsou skupinou poruch způsobených nefunkčními mitochondrie, organely které generují energii pro buňku. Mitochondrie se nacházejí ve všech buňkách lidského těla kromě červené krvinky a přeměňují energii molekul potravin na ATP který napájí většinu funkcí buňky.
Mitochondriální nemoci získávají jedinečné vlastnosti jednak kvůli způsobu, jakým jsou nemoci často dědičné, jednak proto, že mitochondrie jsou tak důležité pro funkci buněk. Podtřída těchto nemocí, které mají neuromuskulární příznaky jsou někdy nazývány mitochondriální myopatie.
Příznaky a symptomy
Mezi příznaky patří:
- špatný růst
- ztráta svalové koordinace
- svalová slabost
- vizuální problémy
- problémy se sluchem
- poruchy učení
- srdeční choroba
- nemoc jater
- nemoc ledvin
- gastrointestinální poruchy
- respirační poruchy
- neurologické problémy
- autonomní dysfunkce
- demence[1]
Získané stavy, ve kterých byla zahrnuta mitochondriální dysfunkce, jsou:
- cukrovka
- Huntingtonova choroba
- rakovina
- Alzheimerova choroba
- Parkinsonova choroba
- bipolární porucha,[2][3][4] schizofrenie, stárnutí a stárnutí, úzkostné poruchy[5]
- kardiovaskulární onemocnění
- sarkopenie
- syndrom chronické únavy[3]
Tělo a každá mutace je modulována jinými variantami genomu; mutace, která u jednoho jedince může způsobit onemocnění jater, může u jiné osoby způsobit poruchu mozku. Závažnost konkrétní vady může být také velká nebo malá. Některé vady zahrnují nesnášenlivost cvičení. Vady často vážněji ovlivňují činnost mitochondrií a více tkání, což vede k multisystémovým onemocněním.[6]
Mitochondriální onemocnění jsou zpravidla horší, pokud se v nich vyskytují defektní mitochondrie svaly, mozek nebo nervy,[7] protože tyto buňky spotřebovávají více energie než většina ostatních buněk v těle.
Přestože se mitochondriální onemocnění od člověka k člověku velmi liší, bylo definováno několik hlavních klinických kategorií těchto stavů na základě nejběžnějších fenotypových znaků, symptomů a známek souvisejících s konkrétními mutacemi, které je obvykle způsobují.[Citace je zapotřebí ]
Vynikající otázkou a oblastí výzkumu je, zda vyčerpání ATP nebo reaktivní formy kyslíku jsou ve skutečnosti zodpovědné za pozorované fenotypové důsledky.[Citace je zapotřebí ]
Cerebelární atrofie nebo hypoplázie bylo někdy údajně spojeno.[8]
Příčiny
Mitochondriální poruchy mohou být způsobeny mutace (získané nebo zděděné) v mitochondriální DNA (mtDNA ) nebo v jaderné geny ten kód pro mitochondriální komponenty. Mohou být také výsledkem získané mitochondriální dysfunkce způsobené nežádoucími účinky léky, infekce nebo jiné příčiny prostředí.[9]
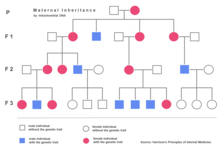
Jaderná DNA má dvě kopie na buňku (s výjimkou spermií a vaječných buněk), přičemž jedna kopie je zděděna po otci a druhá po matce. Mitochondriální DNA se však dědí pouze od matky (s některé výjimky ) a každý mitochondriální organela obvykle obsahuje mezi 2 a 10 mtDNA kopie. V době buněčné dělení mitochondrie se náhodně segregují mezi dvěma novými buňkami. Tyto mitochondrie vytvářejí více kopií a obvykle dosahují 500 mitochondrií na buňku. Když se mtDNA kopíruje, když mitochondrie proliferují, mohou akumulovat náhodné mutace, což je jev nazývaný heteroplazmy. Pokud je vadných pouze několik kopií mtDNA zděděných od matky, mitochondriální dělení může způsobit, že většina defektních kopií skončí pouze v jedné z nových mitochondrií (podrobnější vzory dědičnosti viz lidská mitochondriální genetika ). Mitochondriální onemocnění se může klinicky projevit, jakmile počet postižených mitochondrií dosáhne určité úrovně; tento jev se nazývá „prahový výraz ".
Mitochondrie mají mnoho stejných opravných drah DNA jako jádra - ale ne všechny;[10] proto se mutace vyskytují častěji v mitochondriální DNA než v nukleární DNA (viz Míra mutace ). To znamená, že poruchy mitochondriální DNA se mohou objevit spontánně a relativně často. Poruchy enzymů, které řídí mitochondriální replikace DNA (všechny jsou kódovány geny v jaderné DNA) mohou také způsobit mutace mitochondriální DNA.
Většina mitochondriálních funkcí a biogeneze je řízena nukleární DNA. Lidská mitochondriální DNA kóduje 13 proteinů dýchací řetězec, zatímco většina z odhadovaných 1 500 proteinů a složek zaměřených na mitochondrie je nukleárně kódována. Poruchy mitochondriálních genů kódovaných nukleárními buňkami jsou spojeny se stovkami fenotypů klinických chorob včetně anémie, demence, hypertenze, lymfom, retinopatie, záchvaty, a neurodevelopmentální poruchy.[11]
Studie vědců z Yale University (publikovaná v 12. Února 2004, vydání New England Journal of Medicine ) zkoumali úlohu mitochondrií v inzulínové rezistenci u potomků pacientů s diabetem 2. typu.[12]Další studie ukázaly, že mechanismus může zahrnovat přerušení procesu mitochondriální signalizace v buňkách těla (intramyocelulární lipidy ). Studie provedená v Pennington Biomedical Research Center v Baton Rouge v Louisianě[13] ukázalo, že to zase částečně deaktivuje geny, které produkují mitochondrie.
Příklady
Mezi příklady mitochondriálních onemocnění patří:
- Mitochondriální myopatie
- Diabetes mellitus a hluchota (TÁTO)
- tato kombinace v raném věku může být způsobena mitochondriální chorobou
- Diabetes mellitus a hluchota lze najít společně z jiných důvodů
- Leberova dědičná optická neuropatie (LHON)
- vizuální ztráta začínající v mladé dospělosti
- oční porucha charakterizovaná progresivní ztrátou centrálního vidění v důsledku degenerace optických nervů a sítnice
- postihuje 1 z 50 000 lidí ve Finsku
- Leighův syndrom subakutní sklerotizující encefalopatie
- po normálním vývoji onemocnění obvykle začíná pozdě v prvním roce života, i když k nástupu může dojít v dospělosti
- dochází k rychlému poklesu funkce a je poznamenán záchvaty, změnami stavu vědomí, demencí, selháním ventilace
- Neuropatie, ataxie, retinitis pigmentosa a ptóza (NARP)
- progresivní příznaky popsané v akronymu
- demence
- Myoneurogenní gastrointestinální encefalopatie (MNGIE)
- gastrointestinální pseudoobstrukce
- neuropatie
- Myoklonická epilepsie s otrhanými červenými vlákny (MERRF)
- progresivní myoklonická epilepsie
- „Ragged Red Fibers“ jsou shluky nemocných mitochondrií, které se hromadí v ponorcesarcolemmal oblast svalového vlákna a objeví se, když je sval zbarven modifikovaným Gömöri trichromová skvrna
- nízký vzrůst
- ztráta sluchu
- laktátová acidóza
- nesnášenlivost cvičení
- MELAS syndrom
- Syndrom vyčerpání mitochondriální DNA
Podmínky jako Friedreichova ataxie může ovlivnit mitochondrie ale nejsou spojeny s mitochondriálními proteiny.
Mechanismy
Efektivní celková energetická jednotka pro dostupnou tělesnou energii se označuje jako denní glykogen výrobní kapacita,[14][15][16] a používá se k porovnání mitochondriálního výdeje zdravých jedinců s postiženými nebo chronicky zbavenými glykogenem. Tato hodnota se u daného jedince pomalu mění, protože dokončení celého cyklu trvá mezi 18 a 24 měsíci.[15]
Kapacita generování glykogenu je zcela závislá na provozních úrovních a je určována mitochondrie ve všech buňky z Lidské tělo;[17] vztah mezi energie generované mitochondriemi a kapacita glykogenu je velmi volná a je zprostředkována mnoha biochemické cesty.[14] Energetický výdej plně zdravé mitochondriální funkce lze přesně předpovědět složitým teoretickým argumentem, ale tento argument není přímý, protože většina energie je spotřebována mozkem a není snadno měřitelná.
Diagnóza
Mitochondriální onemocnění jsou obvykle detekována analýzou vzorků svalů, kde je přítomnost těchto organel vyšší. Nejběžnějšími testy pro detekci těchto onemocnění jsou:
- Southern blot k detekci velkých výmazů nebo duplikací
- Polymerázová řetězová reakce a konkrétní testování mutací
- Sekvenování
Ošetření
Ačkoli výzkum stále probíhá, možnosti léčby jsou v současné době omezené; vitamíny jsou často předepisovány, i když důkazy o jejich účinnosti jsou omezené.[18] Pyruvát byla v roce 2007 navržena jako možnost léčby.[19] N-acetylcystein obrací mnoho modelů mitochondriální dysfunkce.[20] V případě poruch nálady, konkrétně bipolární porucha, předpokládá se, že N-acetylcystein (NAC), acetyl-L-karnitin (ALCAR), S-adenosylmethionin (SAMe), koenzym Q10 (CoQ10), kyselina alfa-lipoová (ALA), kreatin monohydrát (CM), a melatonin by mohly být potenciální možnosti léčby.[21]
Genová terapie před koncepcí
Přenos vřetena, Kde nukleární DNA je přenesen do jiné zdravé vaječné buňky a zanechává vadný mitochondriální DNA pozadu je potenciální léčebný postup, který byl úspěšně proveden na opicích.[22][23] Použití podobného pronukleární přenos technology, researchers ve společnosti Newcastle University vedené Douglass Turnbull úspěšně transplantovala zdravou DNA do lidských vajec od žen s mitochondriální chorobou do vajec dárkyň, které nebyly ovlivněny.[24][25] V takových případech byly vzneseny etické otázky týkající se biologického mateřství, protože dítě dostává geny a regulační molekuly genů od dvou různých žen. Použití genetického inženýrství při pokusech o produkci dětí bez mitochondriálních onemocnění je v některých kruzích kontroverzní a je důležité etické problémy.[26][27] Mužské dítě se narodilo v Mexiku v roce 2016 matce s Leighovým syndromem pomocí vřetenového přenosu.[28]
V září 2012 byla ve Velké Británii zahájena veřejná konzultace s cílem prozkoumat související etické problémy.[29] Lidské genetické inženýrství bylo použito v malém měřítku, aby umožnilo neplodným ženám s genetickými vadami mitochondrie mít děti.[30] V červnu 2013 se Spojené království vláda souhlasila s vytvořením legislativy, která by legalizovala „tři osoby“ IVF „postup jako léčba fixující nebo eliminující mitochondriální nemoci, které se přenášejí z matky na dítě. Postup by mohl být nabídnut od 29. října 2015, jakmile budou stanovena nařízení.[31][32][33]Embryonální mitochondriální transplantace a ochrana byly navrženy jako možná léčba dědičného mitochondriálního onemocnění a alotopický výraz mitochondriálních proteinů jako radikální léčba zátěže mutací mtDNA.
V červnu 2018 doporučil Výbor pro komunitní záležitosti Senátu australského senátu přechod k legalizaci Mitochondriální substituční terapie (MR T). Na výzkum a klinické aplikace MRT dohlížely zákony federálních a státních vlád. Státní zákony byly z větší části v souladu s federálním zákonem. Ve všech státech legislativa zakazovala používání technik MRT na klinice a kromě Západní Austrálie byl až do 14. dne vývoje embryí přípustný výzkum omezeného rozsahu MRT s výhradou udělení licence. V roce 2010 se Hon. Poslanec Mark Butler, tehdejší spolkový ministr pro duševní zdraví a stárnutí, jmenoval nezávislý výbor, který přezkoumá dva příslušné akty: Zákon o zákazu klonování lidí pro reprodukci z roku 2002 a Zákon o výzkumu lidských embryí z roku 2002. Zpráva výboru vydaná v červenci 2011 doporučila, aby stávající právní předpisy zůstaly nezměněny
V současné době probíhají klinické studie na lidech na společnostech GenSight Biologics (ClinicalTrials.gov # NCT02064569) a University of Miami (ClinicalTrials.gov # NCT02161380) za účelem zkoumání bezpečnosti a účinnosti mitochondriální genové terapie u Leberovy dědičné optické neuropatie.
Epidemiologie
U přibližně 1 ze 4 000 dětí ve Spojených státech se ve věku 10 let rozvine mitochondriální onemocnění. Až 4 000 dětí ročně se v USA narodí s typem mitochondriální nemoci.[34] Protože mitochondriální poruchy obsahují mnoho variací a podmnožin, některé konkrétní mitochondriální poruchy jsou velmi vzácné.
Průměrný počet narozených za rok u žen s rizikem přenosu choroby mtDNA se odhaduje na přibližně 150 v EU Spojené království a 800 v Spojené státy.[35]
Dějiny
První patogenní mutace v mitochondriální DNA byla identifikována v roce 1988; od té doby do roku 2016 bylo identifikováno přibližně 275 dalších mutací způsobujících onemocnění.[36]:37
Pozoruhodné případy
Pozoruhodné osoby, které trpěly mitochondriální chorobou, zahrnují:
- Mattie Štěpánek, básník, obhájce míru a motivační řečník, který trpěl dysautonomickou mitochondriální myopatií a zemřel ve věku 13 let.
- Rocco Baldelli, trenér a bývalý hráč v poli v centru města Major League Baseball který musel odejít z aktivní hry ve věku 29 let kvůli mitochondriální channelopatii.
- Charlie Gard, britský chlapec, který trpěl syndrom vyčerpání mitochondriální DNA; rozhodnutí o jeho péči byla přijímána různými právními soudy.
Reference
- ^ Nenad Blau; Marinus Duran; K. Michael Gibson; Carlo Dionisi Vici (08.07.2014). Průvodce lékaře k diagnostice, léčbě a následnému sledování zděděných metabolických nemocí. Springer. 339–. ISBN 978-3-642-40337-8.
- ^ Stork, C; Renshaw, P F (2005). „Mitochondriální dysfunkce u bipolární poruchy: důkazy z výzkumu magnetické rezonanční spektroskopie“. Molekulární psychiatrie. 10 (10): 900–19. doi:10.1038 / sj.mp.4001711. PMID 16027739.
- ^ A b Pieczenik, Steve R; Neustadt, John (2007). "Mitochondriální dysfunkce a molekulární dráhy nemoci". Experimentální a molekulární patologie. 83 (1): 84–92. doi:10.1016 / j.yexmp.2006.09.008. PMID 17239370.
- ^ Nierenberg, Andrew A; Kansky, Christine; Brennan, Brian P; Shelton, Richard C; Perlis, Roy; Iosifescu, Dan V (2012). „Mitochondriální modulátory pro bipolární poruchu: patofyziologicky poučené paradigma pro vývoj nových léků“. Australian & New Zealand Journal of Psychiatry. 47 (1): 26–42. doi:10.1177/0004867412449303. PMID 22711881.
- ^ Misiewicz, Zuzanna; Iurato, Stella; Kulesskaya, Natalia; Salminen, Laura; Rodrigues, Luis; Maccarrone, Giuseppina; Martins, Jade; Czamara, Darina; Laine, Mikaela A .; Sokolowska, Ewa; Trontti, Kalevi; Rewerts, Christiane; Novak, Bozidar; Volk, Naama; Park, Dong Ik; Jokitalo, Eija; Paulin, Lars; Auvinen, Petri; Voikar, Vootele; Chen, Alon; Erhardt, Angelika; Turck, Christoph W .; Hovatta, Iiris (2019-09-26). „Multi-omická analýza identifikuje mitochondriální cesty spojené s chováním souvisejícím s úzkostí“. Genetika PLOS. 15 (9): e1008358. doi:10.1371 / journal.pgen.1008358. ISSN 1553-7404. PMC 6762065. PMID 31557158.
- ^ Nunnari J, Suomalainen A (2012). „Mitochondrie: v nemoci a ve zdraví“. Buňka. 148 (6): 1145–59. doi:10.1016 / j.cell.2012.02.035. PMC 5381524. PMID 22424226.
- ^ Finsterer, Josef (2007). "Hematologické projevy primárních mitochondriálních poruch". Acta Haematologica. 118 (2): 88–98. doi:10.1159/000105676. PMID 17637511.
- ^ Lax, Nichola Zoe; Hepplewhite, Philippa Denis; Reeve, Amy Katherine; Nesbitt, Victoria; McFarland, Robert; Jaros, Evelyn; Taylor, Robert William; Turnbull, Douglass Matthew (2012). "Cerebelární ataxie u pacientů s onemocněním mitochondriální DNA". Journal of Neuropathology & Experimental Neurology. 71 (2): 148–61. doi:10.1097 / NEN.0b013e318244477d. PMC 3272439. PMID 22249460.
- ^ "Mitochondriální nemoci". Pletivo. Citováno 2. srpna 2019.
- ^ Alexeyev M, Shokolenko I, Wilson G, LeDoux S (květen 2013). „Zachování integrity mitochondriální DNA - kritická analýza a aktualizace“. Perspektivy Cold Spring Harbor v biologii. 5 (5): a012641. doi:10.1101 / cshperspect.a012641. PMC 3632056. PMID 23637283.
- ^ Scharfe C, Lu HH, Neuenburg JK, Allen EA, Li GC, Klopstock T, Cowan TM, Enns GM, Davis RW (2009). Rzhetsky A (ed.). „Mapování genových asociací v lidských mitochondriích pomocí klinických fenotypů onemocnění“. PLOS Comput Biol. 5 (4): e1000374. Bibcode:2009PLSCB ... 5E0374S. doi:10.1371 / journal.pcbi.1000374. PMC 2668170. PMID 19390613.
- ^ Petersen, Kitt Falk; Dufour, Sylvie; Befroy, Douglas; Garcia, Rina; Shulman, Gerald I. (2004). „Zhoršená mitochondriální aktivita u inzulin rezistentních potomků pacientů s diabetem 2. typu“. New England Journal of Medicine. 350 (7): 664–671. doi:10.1056 / NEJMoa031314. ISSN 0028-4793. PMC 2995502. PMID 14960743.
- ^ Cukrovka 54, 2005 1926-33
- ^ A b Mitchell, Peter. „Koncept respiračního řetězce Davida Keilina a jeho chemiosmotické důsledky“ (PDF). Institut Nobelovy ceny.
- ^ A b Michelakis, Evangelos (leden 2007). „Osa kanálu Mitochondria-K + je potlačena u rakoviny a její normalizace podporuje apoptózu a inhibuje růst rakoviny“. University of Alberta. University of Alberta, 2007. 11 (1): 37–51. doi:10.1016 / j.ccr.2006.10.020. PMID 17222789.
- ^ Lorini a Ciman, M a M (1962). "Hypoglykemický účinek diisopropylamoniových solí při experimentálním diabetu". Institute of Biochemistry, University of Padua, September 1962. Biochemická farmakologie. 11 (9): 823–827. doi:10.1016/0006-2952(62)90177-6. PMID 14466716.
- ^ Stacpoole PW, Henderson GN, Yan Z, James MO (1998). "Klinická farmakologie a toxikologie dichloracetátu". Environ. Perspektiva zdraví. 106 Suppl 4: 989–94. doi:10,1289 / ehp.98106s4989. PMC 1533324. PMID 9703483.
- ^ Manželství B, Clandinin MT, Glerum DM (2003). "Nutriční léčba kofaktorem u mitochondriálních poruch". J Am Diet Doc. 103 (8): 1029–38. doi:10.1016 / S0002-8223 (03) 00476-0. PMID 12891154.
- ^ Tanaka M, Nishigaki Y, Fuku N, Ibi T, Sahashi K, Koga Y (2007). "Terapeutický potenciál léčby pyruvátem pro mitochondriální onemocnění". Mitochondrie. 7 (6): 399–401. doi:10.1016 / j.mito.2007.07.002. PMID 17881297.
- ^ Frantz MC, Wipf P (červen 2010). „Mitochondrie jako cíl léčby“. Environ Mol Mutagen. 51 (5): 462–75. doi:10.1002 / em.20554. PMC 2920596. PMID 20175113.
- ^ Nierenberg, Andrew A, Kansky, Christine, Brennan, Brian P, Shelton, Richard C, Perlis, Roy, Iosifescu, Dan V (2012). „Mitochondriální modulátory pro bipolární poruchu: patofyziologicky poučené paradigma pro vývoj nových léků“. Australian & New Zealand Journal of Psychiatry. 47 (1): 26–42. doi:10.1177/0004867412449303. PMID 22711881.
- ^ Genetický pokrok zvyšuje naděje IVF Autor: Pallab Ghosh. BBC News, vědecký korespondent. Stránka byla naposledy aktualizována v 17:04 GMT, středa 26. srpna 2009 18:04 UK
- ^ Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, Woodward J, Li Y, Ramsey C, Kolotushkina O, Mitalipov S (Září 2009). „Náhrada mitochondriálních genů u potomků primátů a embryonálních kmenových buněk“. Příroda. 461 (7262): 367–372. Bibcode:2009Natur.461..367T. doi:10.1038 / nature08368. PMC 2774772. PMID 19710649.
- ^ Boseley, Sarah (14.04.2010). „Vědci odhalují techniku výměny genů, která maří zděděná onemocnění“. Strážce. Londýn.
- ^ Craven, Lyndsey; Tuppen, Helen A .; Greggains, Gareth D .; Harbottle, Stephen J .; Murphy, Julie L .; Cree, Lynsey M .; Murdoch, Alison P .; Chinnery, Patrick F .; Taylor, Robert W .; Lightowlers, Robert N .; Herbert, Mary; Turnbull, Douglass M. (2010). „Pronukleární přenos do lidských embryí, aby se zabránilo přenosu mitochondriální choroby DNA“. Příroda. 465 (7294): 82–85. Bibcode:2010Natur.465 ... 82C. doi:10.1038 / nature08958. PMC 2875160. PMID 20393463.

- ^ „Spojené království naléhalo na povolení postupu IVF k prevenci smrtelných genetických chorob“. Opatrovník. Londýn. 2015-04-30.
- ^ „Zákon o třech rodičích pro děti je„ nezodpovědný “, říká Anglie před hlasováním. The Telegraph. Londýn. 2015-04-30.
- ^ Hamzelou, Jessica (2016-09-27). „Exkluzivní: První dítě na světě narozené novou technikou„ 3 rodičů “. Nový vědec. Citováno 2016-11-26.
- ^ Ukázka, Ian (2012-09-17). „Regulátor konzultuje veřejnost ohledně plánů nových způsobů léčby plodnosti“. Opatrovník. Londýn. Citováno 8. října 2012.
- ^ "Geneticky pozměněné děti narozené". BBC novinky. 2001-05-04. Citováno 2008-04-26.
- ^ Předpisy o lidském oplodnění a embryologii (darování mitochondrií) z roku 2015 č. 572
- ^ „Vláda Spojeného království podporuje IVF pro tři osoby“. BBC novinky. 27. června 2013.
- ^ Knapton, Sarah (1. března 2014) V Británii by se příští rok mohly narodit „tři rodiče“ The Daily Telegraph Science News, Citováno 1. března 2014
- ^ Centrum pro mitochondriální a metabolická onemocnění
- ^ Gorman, Gráinne S .; Grady, John P .; Ng, Yi; Schaefer, Andrew M .; McNally, Richard J .; Chinnery, Patrick F .; Yu-Wai-Man, Patrick; Herbert, Mary; Taylor, Robert W .; McFarland, Robert; Turnbull, Doug M. (2015). „Mitochondriální dar - kolik žen by z toho mohlo mít prospěch?“. New England Journal of Medicine. 372 (9): 885–887. doi:10.1056 / NEJMc1500960. ISSN 0028-4793. PMC 4481295. PMID 25629662.
- ^ Výbor pro otázky etické a sociální politiky nových technik pro prevenci přenosu mitochondriálních chorob DNA na matku; Rada pro politiku ve zdravotnictví; Institute of Medicine (2016). Claiborne, Anne; Angličtina, Rebecca; Kahn, Jeffrey (eds.). Techniky náhrady mitochondrií: etické, sociální a politické aspekty. Národní akademie Press. ISBN 978-0-309-38870-2. Rejstřík s odkazy na souhrny včetně souhrnný leták o jedné stránce.
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |