Wiskott – Aldrichův syndrom - Wiskott–Aldrich syndrome
| Wiskott-Aldrichův syndrom | |
|---|---|
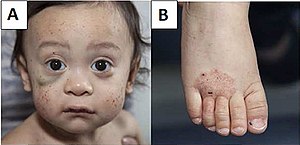 | |
| A) Mnohočetné petechie a hematom pod pravým okem (vlevo na obrázku). B) Ekzém nohy. | |
| Specialita | Imunologie |
Wiskott – Aldrichův syndrom (BOL) je vzácný X-vázaný recesivní nemoc charakterizovaná ekzém, trombocytopenie (nízký destička počet), imunitní nedostatečnost a krvavý průjem (sekundární k trombocytopénii).[1] To je také někdy nazýváno syndrom ekzém-trombocytopenie-imunodeficience v souladu s Aldrichovým původním popisem z roku 1954.[2] Poruchy WAS související s X-vázaná trombocytopenie (XLT) a vrozená neutropenie spojená s X (XLN) mohou vykazovat podobné, ale méně závažné příznaky a jsou způsobeny mutacemi stejného genu.
Příznaky a symptomy
WAS se vyskytuje nejčastěji u mužů kvůli jeho recesivnímu způsobu dědičnosti vázaného na X, který postihuje 1 až 10 mužů na milion.[1] První příznaky jsou obvykle petechie a modřiny vyplývající z nízkého počtu krevních destiček (tj. trombocytopenie ). Spontánní krvácení z nosu a krvavý průjem jsou také běžné a ekzém se obvykle vyvíjí během prvního měsíce života. Opakující se bakteriální infekce rozvíjet o tři měsíce. U většiny dětí s WAS se vyvine alespoň jedno autoimunitní porucha, a rakoviny (hlavně lymfom a leukémie ) se vyvinou až u třetiny pacientů.[3] Imunoglobulin M. Hladiny (IgM) jsou sníženy, IgA a IgE jsou zvýšené a IgG úrovně mohou být normální, snížené nebo zvýšené.[4] Kromě trombocytopenie mají WAS pacienti abnormálně malé krevní destičky (tj. Mikrotrombocyty) a ~ 30% také zvýšené eosinofil počty (tj. eosinofilie ).[5]
Patofyziologie
Mikrotrombocyty pozorované u pacientů s WAS byly pozorovány pouze u jednoho dalšího stavu, ARPC1B nedostatek.[6] V obou případech se předpokládá, že poškozené destičky jsou odstraněny z oběhu pomocí slezina a / nebo játra, což vede k nízkému počtu krevních destiček. Pacienti s WAS měli zvýšenou náchylnost k infekcím, zejména uší a dutin, a tato imunitní nedostatečnost byla spojena se snížením protilátka produkce a neschopnost imunity T buňky účinně bojovat proti infekci.[7]
Genetika

WAS je spojen s mutacemi v a gen na krátkém rameni X chromozom (Xp11.23), který byl původně nazýván Protein Wiskott-Aldrichova syndromu gen a je oficiálně známý jako BOL (ID genu: 7454).[8] Je také spojena s X-vázaná trombocytopenie BOL mutace, i když se liší od těch, které způsobují plnohodnotné WAS. Vzácná porucha X-vázaná neutropenie byla také spojena s konkrétní podskupinou BOL mutace.[9]
Bílkovinný produkt BOL je známý jako WASp. Obsahuje 502 aminokyseliny a je vyjádřena hlavně v krvetvorné buňky (buňky v kostní dřeni, které se vyvinou do krvinek). Hlavní funkcí WASp je aktivace aktinové polymerace tím, že slouží jako faktor podporující tvorbu jader (NPF) pro Komplex Arp2 / 3, který generuje rozvětvená aktinová vlákna. Několik proteinů může sloužit jako NPF a bylo pozorováno, že v destičkách WAS funguje komplex Arp2 / 3 normálně, což naznačuje, že WASp není vyžadován pro svou aktivaci v destičkách.[10] V T-buňkách je WASp důležitý, protože je známo, že je aktivován prostřednictvím Receptor T-buněk signální dráhy k indukci kortikálního aktinu cytoskelet přeskupení, která jsou odpovědná za formování imunologická synapse.[11]
Závažnost příznaků vyvolaných BOL mutace korelují s jejich účinky na WASp. Alely, které neprodukují žádný nebo zkrácený protein, mají závažnější účinky než missense mutace.[12] I když u obou typů mutací může dojít k autoimunitnímu onemocnění a malignitě, u pacientů se zkrácením Vosa nést vyšší riziko. Závada v CD43 molekula byla také nalezena u pacientů s WAS.[13]
Diagnóza
Diagnóza je stanovena na základě klinických parametrů, nátěr periferní krve a nízko imunoglobulin úrovně. Typicky jsou hladiny IgM nízké, hladiny IgA jsou zvýšené a hladiny IgE mohou být zvýšené; paraproteiny jsou občas pozorovány.[14] Hyposenzitivita může odhalit kožní imunologické testování (testování na alergie). Ne všichni pacienti mají pozitivní rodinnou anamnézu poruchy; dochází k novým mutacím. Často je možné podezření na leukémii na základě nízkých krevních destiček a infekcí a biopsie kostní dřeně může být provedeno. Snížené hladiny WASp jsou obvykle pozorovány. Současný zlatý standard pro diagnostiku je genomický Sekvenční analýza DNA, který dokáže detekovat WAS a související poruchy XLT a XLN u velké většiny pacientů a nosičů.[Citace je zapotřebí ]
Klasifikace
Jin a kol. (2004) používají numerické hodnocení závažnosti:[12]
- 0,5: přerušovaná trombocytopenie
- 1.0: trombocytopenie a malé destičky (mikrotrombocytopenie)
- 2.0: mikrotrombocytopenie plus normálně reagující ekzém nebo příležitostné infekce horních cest dýchacích
- 2.5: mikrotrombocytopenie a na léčbu reagující, ale závažné ekzémy nebo infekce dýchacích cest vyžadující antibiotika
- 3.0: mikrotrombocytopenie plus ekzém i infekce dýchacích cest vyžadující antibiotika
- 4.0: mikrotrombocytopenie plus ekzém trvale vyžadující terapii a / nebo závažné nebo život ohrožující infekce
- 5.0: mikrotrombocytopenie plus autoimunitní onemocnění nebo malignita
Léčba
Léčba Wiskott – Aldrichova syndromu je v současné době založena na úpravě příznaků. Aspirin a další nesteroidní protizánětlivé léky je třeba se vyvarovat, protože by mohly narušit funkci destiček, která je již narušena. Ochrannou přilbu můžete chránit před dětmi krvácení do mozku které by mohly být důsledkem poranění hlavy. U pacientů s velmi nízkým počtem krevních destiček mohou pacienti vyžadovat transfuzi krevních destiček nebo odstranění sleziny. U pacientů s častými infekcemi intravenózní imunoglobuliny (IVIG) lze podávat k posílení imunitního systému. Anémie z krvácení může vyžadovat doplnění železa nebo krevní transfúze.[Citace je zapotřebí ]
Jelikož WAS je primárně porucha tkání tvořících krev, a krvetvorné kmenová buňka transplantace, provedená prostřednictvím pupečníková krev nebo transplantace kostní dřeně nabízí jedinou současnou naději na vyléčení. To může být doporučeno u pacientů s HLA -identičtí dárci, shodní sourozenečtí dárci nebo dokonce v případě neúplných shod, pokud je pacientovi věk do 5 let.[Citace je zapotřebí ]
Studie korekce Wiskott – Aldrichova syndromu pomocí genová terapie používat lentivirus začal.[15][16]Pro pacienty s Wiskott-Aldrichovým syndromem byl poskytnut důkaz principu úspěšné genové terapie hematopoetických kmenových buněk.[17]V současné době mnoho vyšetřovatelů pokračuje ve vývoji optimalizovaných vektorů genové terapie.[12][15][16][18] V červenci 2013 italský Institut pro genovou terapii San Raffaele Telethon (HSR-TIGET) uvedl, že tři děti s Wiskott-Aldrichovým syndromem vykazovaly 20–30 měsíců po léčbě geneticky modifikovaným lentivirem výrazné zlepšení.[19] V dubnu 2015 byly výsledky slibné popsány v následném britském a francouzském hodnocení, kde bylo šest dětí s Wiskott – Aldrichovým syndromem léčeno genovou terapií.[20][21] Medián doby sledování byl 27 měsíců.
Prognóza
Výsledky Wiskott-Aldrichova syndromu jsou velmi variabilní v závislosti na tom, jak vážně je postižen jedinec, s mírným koncem spektra nemoci spojeným s BOL gen označovaný jako X-vázaná trombocytopenie nebo X-vázaná neutropenie. Wiskott – Aldrichův syndrom je spojován s variabilní expresivita, což znamená, že dokonce i ve stejné rodině mohou někteří vykazovat pouze chronickou trombocytopenii, zatímco u jiných se v dětství nebo v dětství vyskytnou závažné život ohrožující komplikace Wiskott-Aldrichova syndromu.[22][23] Vzhledem k tomu, že příznaky často s věkem postupují, je obtížné předvídat, jak vážně bude nově diagnostikované dítě nakonec ovlivněno. Životnost trombocytopenie vázané na X je považována za normální, zprávy o minimálně postižených mužích v sedmém desetiletí.[24] Existuje určitá korelace genotyp-fenotyp, přičemž většina jedinců má trombocytopenii spojenou s X varianty missense v BOL gen oproti 86,5% těch, kteří nevytvářejí WASp, mají fenotyp syndromu Wiskott – Aldrich.[25] Celková prognóza u jedinců s Wiskott-Aldrichovým syndromem se za posledních několik desetiletí výrazně zlepšila díky dřívějším diagnózám a většímu přístupu k možnostem léčby, jako je transplantace hematopoetických kmenových buněk.
Epidemiologie
Odhadovaný výskyt Wiskott-Aldrichova syndromu ve Spojených státech je jeden z 250 000 živě narozených mužů. Neexistuje žádný geografický faktor.[26]
Dějiny
Tento syndrom je pojmenován po Dr. Alfredu Wiskottovi (1898–1978), německém pediatrovi, který si syndrom poprvé všiml v roce 1937,[27] a Dr. Robert Anderson Aldrich (1917–1998), americký pediatr, který tuto chorobu popsal v rodině nizozemských Američanů v roce 1954.[2] Wiskott popsal tři bratry s podobnou nemocí, jejichž sestry nebyly ovlivněny. V roce 2006 německá výzkumná skupina analyzovala rodinné příslušníky Wiskottových tří případů a domnívala se, že pravděpodobně sdílejí novou mutaci posunu prvního exonu Vosa gen.[28]
Reference
- ^ A b Reference, Genetics Home. "Wiskott-Aldrichův syndrom". Genetická domácí reference. Citováno 2016-06-26.
- ^ A b Aldrich RA, Steinberg AG, Campbell DC (únor 1954). „Rodokmen prokazující recesivní stav spojený s pohlavím charakterizovaný vyčerpáním uší, ekzematoidní dermatitidou a krvavým průjmem“. Pediatrie. 13 (2): 133–9. PMID 13133561.
- ^ Wiskott-Aldrichův syndrom na eMedicína
- ^ Sande MA, Wilson WP (2001). Současná diagnostika a léčba infekčních nemocí. New York: Lékařské knihy Lange / McGraw-Hill. str. 361. ISBN 978-0-8385-1494-8.[stránka potřebná ]
- ^ Navabi B, Upton JE (2016). „Primární imunodeficience spojené s eozinofilií“. Alergie, astma a klinická imunologie. 12: 27. doi:10.1186 / s13223-016-0130-4. PMC 4878059. PMID 27222657.
- ^ Kahr WH, Pluthero FG, Elkadri A, Warner N, Drobac M, Chen CH, Lo RW, Li L, Li R, Li Q, Thoeni C, Pan J, Leung G, Lara-Corrales I, Murchie R, Cutz E, Laxer RM, Upton J, Roifman CM, Yeung RS, Brumell JH, Muise AM (duben 2017). „Ztráta komplexní složky Arp2 / 3 ARPC1B způsobuje abnormality krevních destiček a předisponuje k zánětlivému onemocnění“. Příroda komunikace. 8: 14816. Bibcode:2017NatCo ... 814816K. doi:10.1038 / ncomms14816. PMC 5382316. PMID 28368018.
- ^ „Wiskott-Aldrichův syndrom: Poruchy imunodeficience: Merck Manual Professional“. Citováno 2008-03-01.
- ^ Derry JM, Ochs HD, Francke U (srpen 1994). "Izolace nového genu mutovaného ve Wiskott-Aldrichově syndromu". Buňka. 78 (4): 635–44. doi:10.1016/0092-8674(94)90528-2. PMID 8069912. S2CID 9040376.
- ^ Westerberg LS, Meelu P, Baptista M, Eston MA, Adamovich DA, Cotta-de-Almeida V, Seed B, Rosen MK, Vandenberghe P, Thrasher AJ, Klein C, Alt FW, Snapper SB (červen 2010). „Aktivace mutací WASP spojených s neutropenií vázanou na X má za následek zvýšenou polymeraci aktinů, změnu cytoskeletálních odpovědí a genomovou nestabilitu v lymfocytech.“. The Journal of Experimental Medicine. 207 (6): 1145–52. doi:10.1084 / jem.20091245. PMC 2882832. PMID 20513746.
- ^ Falet H, Hoffmeister KM, Neujahr R, Hartwig JH (září 2002). „Normální aktivace komplexu Arp2 / 3 v destičkách bez WASp“. Krev. 100 (6): 2113–22. doi:10.1182 / krev.V100.6.2113. PMID 12200375.
- ^ Malinova D, Fritzsche M, Nowosad CR, Armer H, Munro PM, Blundell MP, Charras G, Tolar P, Bouma G, Thrasher AJ (květen 2016). „Stabilita aktinového cytoskeletu závislého na WASp v imunologické synapse dendritických buněk je vyžadována pro rozsáhlé funkční kontakty T buněk“. Journal of Leukocyte Biology. 99 (5): 699–710. doi:10.1189 / jlb.2a0215-050rr. PMC 5404712. PMID 26590149.
- ^ A b C Jin Y, Mazza C, Christie JR, Giliani S, Fiorini M, Mella P, Gandellini F, Stewart DM, Zhu Q, Nelson DL, Notarangelo LD, Ochs HD (prosinec 2004). „Mutace proteinu Wiskott-Aldrichova syndromu (WASP): aktivní body, účinek na transkripci a translaci a korelaci fenotypu / genotypu“. Krev. 104 (13): 4010–9. doi:10.1182 / krev-2003-05-1592. PMID 15284122.
- ^ Rosenstein Y, Park JK, Hahn WC, Rosen FS, Bierer BE, Burakoff SJ (listopad 1991). „CD43, molekula defektní při Wiskott-Aldrichově syndromu, váže ICAM-1“. Příroda. 354 (6350): 233–5. Bibcode:1991 Natur.354..233R. doi:10.1038 / 354233a0. PMID 1683685. S2CID 4256329.
- ^ Radl J, Dooren LH, Morell A, Skvaril F, Vossen JM, Uittenbogaart CH (srpen 1976). „Imunoglobuliny a přechodné paraproteiny v séru pacientů se syndromem Wiskott-Aldrich: následná studie“. Klinická a experimentální imunologie. 25 (2): 256–63. PMC 1541349. PMID 954233.
- ^ A b Galy A, Roncarolo MG, Thrasher AJ (únor 2008). „Vývoj lentivirové genové terapie pro syndrom Wiskotta Aldricha“. Znalecký posudek na biologickou terapii. 8 (2): 181–90. doi:10.1517/14712598.8.2.181. PMC 2789278. PMID 18194074.
- ^ A b Frecha C, Toscano MG, Costa C, Saez-Lara MJ, Cosset FL, Verhoeyen E, Martin F (červen 2008). „Vylepšené lentivirové vektory pro genovou terapii Wiskott-Aldrichova syndromu napodobují profily endogenní exprese během hematopoézy“. Genová terapie. 15 (12): 930–41. doi:10.1038 / gt.2008.20. PMID 18323794.
- ^ Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Díez IA, Dewey RA, Böhm M, Nowrouzi A, Ball CR, Glimm H, Naundorf S, Kühlcke K, Blasczyk R, Kondratenko I, Maródi L, Orange JS, von Kalle C, Klein C (listopad 2010). „Genová terapie kmenovými buňkami pro Wiskott-Aldrichův syndrom“. The New England Journal of Medicine. 363 (20): 1918–27. doi:10.1056 / NEJMoa1003548. PMC 3064520. PMID 21067383.
- ^ Dewey RA, Avedillo Díez I, Ballmaier M, Filipovich A, Greil J, Güngör T, Happel C, Maschan A, Noyan F, Pannicke U, Schwarz K, Snapper S, Welte K, Klein C (září 2006). „Retrovirový přenos genu WASP do lidských hematopoetických kmenových buněk rekonstituuje aktinový cytoskelet v myeloidních potomcích diferencovaných in vitro.“ Experimentální hematologie. 34 (9): 1161–9. doi:10.1016 / j.exphem.2006.04.021. PMID 16939809.
- ^ Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC, Bosticardo M, Evangelio C, Assanelli A, Casiraghi M, Di Nunzio S, Callegaro L, Benati C, Rizzardi P, Pellin D, Di Serio C, Schmidt M, Von Kalle C, Gardner J, Mehta N, Neduva V, Dow DJ, Galy A, Miniero R, Finocchi A, Metin A, Banerjee PP, Orange JS, Galimberti S, Valsecchi MG, Biffi A, Montini E, Villa A, Ciceri F, Roncarolo MG, Naldini L (srpen 2013). „Genetická terapie lentivirovými hematopoetickými kmenovými buňkami u pacientů s Wiskott-Aldrichovým syndromem“. Věda. 341 (6148): 1233151. doi:10.1126 / science.1233151. PMC 4375961. PMID 23845947.
- ^ Gallagher, James (21. dubna 2015) Genová terapie: „Zkrotit HIV“ se používá k léčbě nemoci BBC News, Health, Citováno 21. dubna 2015
- ^ Malech HL, Ochs HD (duben 2015). „Nastávající éra klinického přínosu z genové terapie“. JAMA. 313 (15): 1522–3. doi:10.1001 / jama.2015.2055. PMID 25898049.
- ^ Buchbinder, David; Nadeau, Kari; Nugent, Diane (2011-06-28). „Monozygotní dvojice dvojčat vykazující nesourodý fenotyp X-vázané trombocytopenie a Wiskott – Aldrichova syndromu: role epigenetiky?“. Journal of Clinical Immunology. 31 (5): 773–777. doi:10.1007 / s10875-011-9561-3. ISSN 0271-9142.
- ^ Albert, Michael H; Notarangelo, Luigi D; Ochs, Hans D (leden 2011). „Klinické spektrum, patofyziologie a léčba Wiskott – Aldrichova syndromu“. Aktuální názor na hematologii. 18 (1): 42–48. doi:10.1097 / moh.0b013e32834114bc. ISSN 1065-6251.
- ^ Albert, Michael H .; Bittner, Tanja C .; Nonoyama, Shigeaki; Notarangelo, Lucia Dora; Burns, Siobhan; Imai, Kohsuke; Espanol, Teresa; Fasth, Anders; Pellier, Isabelle; Strauss, Gabriele; Morio, Tomohiro (2010-04-22). „Trombocytopenie vázaná na X (XLT) způsobená mutacemi WAS: klinické charakteristiky, dlouhodobý výsledek a možnosti léčby“. Krev. 115 (16): 3231–3238. doi:10.1182 / krev-2009-09-239087. ISSN 0006-4971.
- ^ Imai, Kohsuke; Nonoyama, Shigeaki; Ochs, Hans D. (prosinec 2003). „Genové mutace a fenotyp WASP (Wiskott-Aldrich syndrom protein). Aktuální názor na alergii a klinickou imunologii. 3 (6): 427–436. doi:10.1097/00130832-200312000-00003. ISSN 1528-4050.
- ^ Robert A Schwartz, MD, MPH; Hlavní redaktor: Harumi Jyonouchi, MD. Dětský Wiskott-Aldrichův syndrom
- ^ Wiskott, A (1937). "Familiärer, angeborener Morbus Werlhofii? („Rodinná vrozená Werlhofova choroba?“) “. Montsschr Kinderheilkd. 68: 212–16.
- ^ Binder V, Albert MH, Kabus M, Bertone M, Meindl A, Belohradsky BH (říjen 2006). „Genotyp původního Wiskottova fenotypu“. The New England Journal of Medicine. 355 (17): 1790–3. doi:10.1056 / NEJMoa062520. PMID 17065640. S2CID 30040802.
Další čtení
- GeneReviews / NIH / NCBI / UW záznam o poruchách souvisejících s WAS, včetně syndromu Wiskott-Aldrich WAS X-vázaná trombocytopenie XLT a X-vázaná vrozená neutropenie XLN
- Nadace pro imunitní nedostatečnost - Kapitola VII, „Wiskott-Aldrichův syndrom“
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |