Von Hippel – Lindauova choroba - Von Hippel–Lindau disease - Wikipedia
| Von Hippel – Lindauova choroba | |
|---|---|
| Ostatní jména | Familiární cerebello retinální angiomatóza[1] |
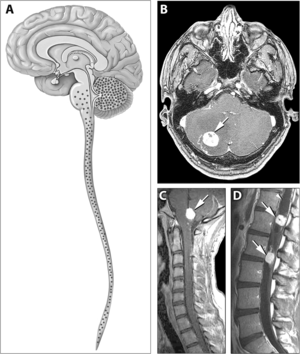 | |
| Typická distribuce hemangioblastomů u Von Hippel – Lindauovy choroby | |
| Specialita | Lékařská genetika, neurologie |
Von Hippel – Lindauova choroba (VHL), také známý jako Von Hippel-Lindauův syndrom, je vzácný genetická porucha s zapojením více systémů.[2] Je charakterizován viscerálními cystami a benigní nádory s potenciálem pro následnou maligní transformaci. Je to typ phakomatosis který je výsledkem a mutace v Von Hippel – Lindau supresor nádoru gen zapnutý chromozom 3p 25.3.[3][4][5]
Příznaky a symptomy
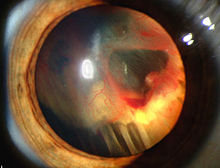
Známky a příznaky spojené s onemocněním VHL zahrnují bolesti hlavy, problémy s rovnováhou a chůzí, závratě, slabost končetin, problémy se zrakem a vysoký krevní tlak. Stavy spojené s onemocněním VHL zahrnují angiomatóza, hemangioblastomy, feochromocytom, karcinom ledvin, pankreatické cysty (pankreatický serózní cystadenom ), nádor endolymfatického vaku a bilaterální papilární cystadenomy nadvarlete (muži) nebo široké vazivo dělohy (ženy).[6][7] Angiomatóza se vyskytuje u 37,2% pacientů s onemocněním VHL a obvykle se vyskytuje v sítnici. Výsledkem je velmi častá ztráta zraku. Mohou však být ovlivněny i jiné orgány: mrtvice, infarkty a kardiovaskulární onemocnění jsou dalšími běžnými příznaky.[5] Přibližně 40% onemocnění VHL vykazuje hemangioblastomy CNS a jsou přítomny přibližně u 60–80%. Páteřní hemangioblastomy se vyskytují u 13-59% onemocnění VHL a jsou specifické, protože 80% se vyskytuje u onemocnění VHL.[8][9] Ačkoli jsou všechny tyto nádory běžné u onemocnění VHL, přibližně u poloviny případů se vyskytuje pouze jeden typ nádoru.[9]
Patogeneze
Toto onemocnění je způsobeno mutacemi Von Hippel – Lindau supresor nádoru (VHL) gen na krátkém rameni chromozom 3 (3p25-26). Existuje více než 1500 zárodečné mutace a somatické mutace nalezený v nemoci VHL.[10][11]

Každá buňka v těle má 2 kopie každého genu (kromě těch, které se nacházejí v pohlavních chromozomech X a Y). U onemocnění VHL má jedna kopie genu VHL mutaci a produkuje vadný protein VHL (pVHL). Druhá kopie však stále produkuje funkční protein. Tento stav je zděděn autozomálně dominantním způsobem - jedna kopie vadného genu je dostatečná ke zvýšení rizika vzniku nádorů. [12][13]
Přibližně 20% případů onemocnění VHL se vyskytuje u jedinců bez rodinné anamnézy, známých jako de novo mutace. Zděděná mutace genu VHL je zodpovědná za zbývajících 80 procent případů.[8]
30-40% mutací v genu VHL sestává z 50-250kb deleční mutace které odstraňují buď část genu, nebo celý gen a přilehlé oblasti DNA. Zbývajících 60–70% onemocnění VHL je způsobeno zkrácením pVHL o nesmyslné mutace, indel mutace nebo mutace místa sestřihu.[8]
VHL protein

Protein VHL (pVHL) se podílí na regulaci proteinu známého jako hypoxií indukovatelný faktor 1α (HIF1α). Toto je podjednotka a heterodimerní transkripční faktor že za normálních okolností buněčný kyslík úrovně je vysoce regulováno. Za normálních fyziologických podmínek pVHL rozpoznává a váže se na HIF1α, pouze pokud je přítomen kyslík po překladu hydroxylace ze dne 2 prolin zbytky v proteinu HIF1α. pVHL je E3 ligáza že ubikvitináty HIF1α a způsobuje jeho degradaci proteazom. V podmínkách s nízkým obsahem kyslíku nebo v případech onemocnění VHL, kde je mutován gen VHL, se pVHL neváže na HIF1α. To umožňuje podjednotce dimerizovat s HIF1β a aktivovat transkripci řady genů, včetně vaskulární endoteliální růstový faktor, růstový faktor B odvozený z krevních destiček, erytropoetin a geny podílející se na absorpci a metabolismu glukózy.[13][14] Nová nová missense mutace v genech VHL c.194 C> T, c.239 G> A, c.278 G> A, c.319 C> G, c.337 C> G vedoucí k následujícím variantám p.Ala 65 Val, p.Gly 80 Asp, p.Gly 93 Glu, p.Gln 107 Glu, p.Gln 113 Glu[kontrolovat pravopis ] v proteinu přispělo k renálnímu karcinomu jasných buněk.[15]
Diagnóza
Detekce nádorů specifických pro onemocnění VHL je důležitá při diagnostice onemocnění. U jedinců s rodinnou anamnézou onemocnění VHL, jeden hemangioblastom, feochromocytom nebo karcinom ledvin může stačit k stanovení diagnózy. Protože všechny nádory spojené s onemocněním VHL lze nalézt sporadicky, je třeba identifikovat alespoň dva nádory, aby bylo možné diagnostikovat onemocnění VHL u osoby bez rodinné anamnézy.[8][9]
Genetická diagnóza je také užitečná při diagnostice onemocnění VHL. U dědičných onemocnění VHL se používají techniky jako Southern blot a genové sekvenování lze použít k analýze DNA a identifikaci mutací. Tyto testy lze použít k screeningu členů rodiny těch, kteří trpí onemocněním VHL; de novo případy, které produkují genetický mozaicismus je obtížnější zjistit, protože mutace se nenacházejí v bílých krvinkách, které se používají pro genetickou analýzu.[8][16]
Klasifikace
Onemocnění VHL lze rozdělit podle klinických projevů, i když tyto skupiny často korelují s určitými typy mutací přítomných v genu VHL.[17]
Léčba
Včasné rozpoznání a léčba specifických projevů VHL může podstatně snížit komplikace a zlepšit kvalitu života. Z tohoto důvodu jsou jedinci s onemocněním VHL obvykle rutinně vyšetřováni na retinální angiómy, hemangioblastomy CNS, renální karcinomy z čirých buněk a feochromocytomy.[18] Hemangioblastomy CNS jsou obvykle chirurgicky odstraněny, pokud jsou symptomatické. Fotokoagulace a kryoterapie se obvykle používají k léčbě symptomatických anginů sítnice, ačkoli alternativou může být i antiangiogenní léčba. Renální nádory lze odstranit částečným nefrektomie nebo jiné techniky, jako je radiofrekvenční ablace.[8]
Epidemiologie
Onemocnění VHL má výskyt jednoho z 36 000 porodů. Existuje více než 90% pronikavost ve věku 65 let.[19] Věk při stanovení diagnózy se liší od kojeneckého věku do věku 60–70 let, přičemž průměrný věk pacienta při klinické diagnóze je 26 let.[Citace je zapotřebí ]
Dějiny

Německý oftalmolog Eugen von Hippel poprvé popsal angiómy v oku v roce 1904.[20] Arvid Lindau popsal angiómy mozeček a páteř v roce 1927.[21] Termín Von Hippel – Lindauova choroba byl poprvé použit v roce 1936; jeho použití se však stalo běžným až v 70. letech.[8]
Pozoruhodné případy
Někteří potomci rodiny McCoy (podílející se na Hatfield-McCoy spor z Appalachia, USA) se předpokládá, že mají VHL. V článku, který vyšel v Associated Press, se spekulovalo endokrinologem z Vanderbiltovy univerzity, že nepřátelství, které je základem sváru Hatfield-McCoy, mohlo být částečně způsobeno následky Von Hippel-Lindauovy choroby. Článek naznačuje, že rodina McCoyů byla náchylná ke špatné náladě, protože mnoho z nich mělo feochromocytom, který produkoval nadměrný adrenalin a sklon k výbušným náladám.[22]
Nomenklatura
Další neobvyklá jména jsou: angiomatóza sítnice, familiární cerebello-retinální angiomatóza, cerebelloretinální hemangioblastomatóza, Hippelova choroba, Hippel-Lindauův syndrom, HLS, VHL, Lindauova choroba nebo retinocerebelární angiomatóza.[23][24]
Viz také
Reference
- ^ VYHRAZENO, VLOŽTE NÁS 14-- VŠECHNA PRÁVA. „Orphanet: Von Hippel Lindau disease“. www.orpha.net. Citováno 25. května 2019.
- ^ "Von Hippel-Lindauova choroba | Informační centrum o genetických a vzácných onemocněních (GARD) - program NCATS". rarediseases.info.nih.gov. Citováno 2018-04-17.
- ^ Richard, S; Gardie, B; Couvé, S; Gad, S (30. května 2012). „Von Hippel-Lindau: Jak vzácné onemocnění osvětluje biologii rakoviny“ (PDF). Semináře z biologie rakoviny. 23 (1): 26–37. doi:10.1016 / j.semcancer.2012.05.005. PMID 22659535.
- ^ Henry, Todd; Campell, James; Hawley, Arthur (1969). Todd-Sanfordova klinická diagnóza laboratorními metodami, editoval Israel Davidsohn [a] John Bernard Henry (14. vydání). Philadelphia: Saunders. p. 555. ISBN 978-0-7216-2921-6.
- ^ A b Wong WT; Agrón E; Coleman HR; et al. (Únor 2007). „Korelace genotyp – fenotyp u von Hippel – Lindauovy choroby s angiomatózou sítnice“. Archiv oftalmologie. 125 (2): 239–45. doi:10.1001 / archopht.125.2.239. PMC 3019103. PMID 17296901. Archivovány od originál dne 12. 12. 2008. Citováno 2008-10-22.
- ^ Lindsay, Kenneth W; Ian Bone; Robin Callander; J. van Gijn (1991). Ilustrovaná neurologie a neurochirurgie. USA: Churchill Livingstone. ISBN 978-0-443-04345-1.
- ^ Frantzen, Carlijn; Odkazy, Thera P .; Giles, Rachel H. (21. června 2012). "Von Hippel-Lindauův syndrom". Von Hippel-Lindauova choroba. GeneReviews v NCBI. University of Washington, Seattle. Citováno 30. března 2013.
- ^ A b C d E F G Maher ER; Glenn GM; Walther M; et al. (Červen 2011). „von Hippel-Lindauova choroba: klinický a vědecký přehled“. European Journal of Human Genetics. 19 (6): 617–23. doi:10.1038 / ejhg.2010.175. PMC 3110036. PMID 21386872.
- ^ A b C Friedrich, CA (1. prosince 1999). "Von Hippel-Lindauův syndrom. Pleomorfní stav". Rakovina. 86 (11 doplňků): 2478–82. doi:10.1002 / (SICI) 1097-0142 (19991201) 86: 11+ <2478 :: AID-CNCR4> 3.0.CO; 2-5. PMID 10630173.
- ^ Kondo, K; Kaelin Jr, WG (10. března 2001). „Gen pro potlačování nádorů von Hippel – Lindau“. Experimentální výzkum buněk. 264 (1): 117–125. doi:10,1006 / excr. 2000,5139. PMID 11237528.
- ^ Nordstrom-O'Brien M; van der Luijt RB; van Rooijen E; et al. (Květen 2010). „Genetická analýza von Hippel-Lindauovy choroby“. Hučení. Mutat. 31 (5): 521–37. doi:10,1002 / humu.21219. PMID 20151405. S2CID 38910112.
- ^ Knudson, AG (listopad 2001). "Dva genetické zásahy (více či méně) do rakoviny". Nature Reviews Cancer. 1 (2): 157–62. doi:10.1038/35101031. PMID 11905807. S2CID 20201610.
- ^ A b Kaelin, WG (2007). „Von Hippel-Lindauova choroba“. Výroční přehled patologie. 2: 145–73. doi:10.1146 / annurev.pathol.2.010506.092049. PMID 18039096.
- ^ Bader, HL; Hsu, T (4. června 2012). „Systémové funkce VHL genu a onemocnění VHL“. FEBS Dopisy. 586 (11): 1562–9. doi:10.1016 / j.febslet.2012.04.032. PMC 3372859. PMID 22673568.
- ^ Kumar, P. S .; Venkatesh, K .; Srikanth, L .; Sarma, P. V .; Reddy, A. R .; Subramanian, S .; Phaneendra, B. V. (červenec 2013). "Nové tři missense mutace pozorované v genu Von Hippel-Lindau u pacienta hlášeného s karcinomem ledvinových buněk". Indian Journal of Human Genetics. 19 (3): 373–376. doi:10.4103/0971-6866.120809. PMC 3841571. PMID 24339559.
- ^ Lonser RR (červen 2003). „von Hippel-Lindauova choroba“. Lanceta. 361 (9374): 2059–67. doi:10.1016 / S0140-6736 (03) 13643-4. PMID 12814730. S2CID 13783714.
- ^ Calzada, MJ (březen 2010). „Von Hippel-Lindauův syndrom: molekulární mechanismy nemoci“. Klinická a translační onkologie. 12 (3): 160–5. doi:10.1007 / s12094-010-0485-9. PMID 20231120. S2CID 7789108.
- ^ Priesemann M; Davies KM; Perry LA; et al. (2006). „Výhody screeningu u von Hippel-Lindauovy choroby - srovnání morbidity spojené s počátečními tumory u postižených rodičů a dětí“. Horm. Res. 66 (1): 1–5. doi:10.1159/000093008. PMID 16651847. S2CID 29862078.
- ^ Kim, JJ; Rini, BI; Hansel, DE (2010). Von Hippel Lindauův syndrom. Pokroky v experimentální medicíně a biologii. 685. s. 228–49. doi:10.1007/978-1-4419-6448-9_22. ISBN 978-1-4419-6447-2. PMID 20687511.
- ^ Von Hippel E (1904). „Ueber eine sehr seltene Erkrankung der Netzhaut“. Albrecht von Graefes Arch Ophthal. 59: 83–106. doi:10.1007 / bf01994821. S2CID 22425158.
- ^ Lindau A (1927). „Zur Frage der Angiomatosis Retinae und Ihrer Hirncomplikation“. Acta Ophthalmol. 4 (1–2): 193–226. doi:10.1111 / j.1755-3768.1926.tb07786.x. S2CID 73385451.
- ^ "Hatfield – McCoyův spor byl obviňován z „vzteklé“ nemoci". MSNBC.com. 2007-04-05. Archivovány od originál dne 2007-04-07. Citováno 2007-04-05.
- ^ „Národní organizace NORD pro vzácné poruchy“.
- ^ „MeSH (Medical Subject Headings)“. Citováno 08/11/2012. Zkontrolujte hodnoty data v:
| accessdate =(Pomoc)
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |
- GeneReviews / NCBI / NIH / UW vstup na Von Hippel-Lindau syndrom
- Von Hippel – Lindauova choroba (VHL) na NINDS
- Von Hippel – Lindauův syndrom na NLM Genetická domácí reference
- von Hippel – Lindauův syndrom na REFRÉN
- Hippel – Lindauova choroba na Kdo to pojmenoval?
- Online Mendelian Inheritance in Man (OMIM): 608537 (Gen VHL)