Kraniofrontonazální dysplázie - Craniofrontonasal dysplasia
| Kraniofrontonazální dysplázie | |
|---|---|
| Ostatní jména | Kraniofrontonasální dysostóza |
 | |
| Tato podmínka se dědí v X-vázaný dominantní způsob. Na rozdíl od většiny podmínek vázaných na X je však u žen závažnější interakce buňka-buňka mechanismy zahrnující odpovědný gen (EFNB1 ) pokud je přítomen pouze v některých buňkách (mozaika ). | |
| Specialita | Lékařská genetika |
Kraniofrontonazální dysplázie (kraniofrontonasální syndrom, kraniofrontonasální dysostóza, CFND) je velmi vzácný X-vázaný malformační syndrom způsobený mutacemi v genu pro efrin-B1 (EFNB1 ).[1][2] Fenotypová exprese se velmi liší u postižených jedinců, kde jsou častěji a obecně více postiženy ženy než muži.[1][2] Běžné fyzické malformace jsou: kraniosynostóza z koronální šev orbitální hypertelorismus, bifidní nosní špička, suchá kudrnatý zvlněné vlasy, podélné rýhy a / nebo štípání nehtů a asymetrie obličeje.[3][4][5][6]
Diagnóza CFND je určena přítomností mutace v genu EFNB1. Fyzikální vlastnosti mohou hrát podpůrnou roli při stanovení diagnózy.
Léčba je vždy chirurgická a je založena na specifické fenotypové prezentaci každého pacienta.[7]
Prezentace
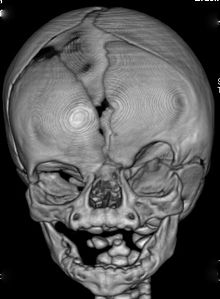
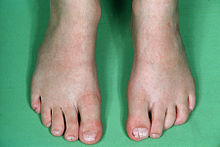
Fenotypová exprese se u jednotlivců s CFND velmi liší. Mezi nejvýznamnější vlastnosti patří:[3][4][5][6]
- Kraniosynostóza z koronální šev (s) (fúze koronálních stehů),
- Orbitální hypertelorismus (zvětšená interokulární vzdálenost),
- Bifidní nosní špička,
- Suché kudrnaté zvlněné vlasy,
- Podélné rýhy a / nebo štípání nehtů,
- Asymetrie obličeje.
Méně často viditelné vlastnosti jsou: široká nosní báze, nízká přední linie vlasů, nízko nasazené uši, vytěsněné zuby, maxilární hypoplázie, zaoblená a šikmá ramena, pectus excavatum, skolióza, vysoké klenuté patro, orbitální dystopie, nízký implantát prsou s asymetrickými bradavkami a objemem, webbed krk, abnormality rukou nebo nohou jako klinodaktylie (nejběžnější je zakřivený 5. prst) a kožní syndactyly (prsty na rukou / nohou).[3][4][5][6]
Ženy jsou častěji a obvykle více postiženy než muži. Muži však mohou mít (některé) stejné příznaky jako ženy, ale to se často neobjevuje.[3] Většina mužů má mírné příznaky, jako je hypertelorismus a široká nosní báze s bifidním nosem, ale může být také nositelem mutace, přesto zůstává klinicky nedotčena.[1][2]
Genetika
CFND je velmi vzácný X-vázaný malformační syndrom způsobený mutacemi v genu pro efrin-B1 (EFNB1 ).[1][2] Gen EFNB1 kóduje membránou ukotvený ligand, který se může vázat na receptor efrin tyrosinkinázy.[2] Tento receptor efrinu je mimo jiné zodpovědný za regulaci formování hranice embryonální tkáně a je důležitý pro vývoj kostry a kraniofaciální oblasti.[8][9] Vzhledem k tomu, že receptor efrinu a jeho ligand EFNB1 jsou oba vázány na (trans) membránu buňky, je jeho kaskáda aktivována prostřednictvím interakcí mezi buňkami.[8] Tyto interakce mezi buňkami jsou narušeny v důsledku přítomnosti buněk s mutantním genem EFNB1, což vede k neúplné tvorbě hranice tkáně.[5]
Paradoxní vůči jiným podmínkám spojeným s X, u CFND jsou ženy vážněji postiženy než muži.[3] To je způsobeno procesem X-inaktivace u žen, kde je náhodně buď mateřský nebo otcovský X-chromozom inaktivován v buňce.[3][10] Díky tomuto procesu obsahují tkáně těla buď buňky s normálním EFNB1, nebo mutovaný EFNB1. Tomu se říká mozaikový vzor.[3][10][11] Tento mozaikový vzor buněk „narušuje“ funkčnost interakcí mezi buňkami, což má za následek závažné fyzické malformace u žen.[11][12]
Stejně jako u všech podmínek vázaných na X má CFND přednastavenou šanci na předání z rodičů na své potomky. Ženy mají dva X-chromozomy a muži mají jeden X-chromozom. Pokud je matka nositelkou CFND, existuje 50% pravděpodobnost, že přenese na své potomky chromozom X obsahující mutovaný gen EFNB1, bez ohledu na to, zda je dítě chlapec nebo dívka. Pokud je otec nosičem, existuje 100% šance, že předá svůj X-chromozom s mutací EFNB1 na dceru a 0% šance, že ho předá synovi.[3]
Diagnóza
Diagnóza CFND je stanovena až po přítomnosti mutace v EFNB1 gen byl stanoven.[1][2][13] Fyzické projevy nemusí být nutně součástí diagnostických kritérií, ale mohou pomoci vést správným směrem. To je způsobeno velkou heterogenitou mezi pacienty, pokud jde o fenotypovou expresi.[7]
20% pacientů, kteří vykazují vlastnosti podobné CFND, nevykazuje mutaci v genu EFNB1.[13][14][15] Skupina pacientů s diagnostikovanou CFND je tak často přeceňována. Je však důležité odlišit tuto populaci od CFND pro výzkumné účely. Na druhou stranu, zejména u mužů, je možné, že někdo je nositelem mutace genu EFNB1, přesto neprojevuje žádné fyzické projevy.[14][15] Screening na přítomnost mutace EFNB1 je tedy nejspolehlivější metodou pro stanovení diagnózy CFND.[Citace je zapotřebí ]
Genetické poradenství nebo prenatální screening může být doporučeno, pokud existuje důvod k podezření na přítomnost mutace genu EFNB1.[3][5] Prenatální screening lze provést provedením ultrazvuk, kde lze konkrétně hledat hypertelorismus nebo bifidní nosní špička. To je však docela obtížné, protože postižení obličeje nemusí být v tak raném věku zřejmé, zejména v případech s mírnou fenotypovou prezentací.[7] Nej definitivnější způsob, jak prokázat přítomnost CFND, je genetické testování amniocentéza a vzorkování choriových klků. To však s sebou nese větší riziko předčasného ukončení těhotenství.[16]
Léčba
Pro lidi s CFND neexistuje žádná „standardní léčba“ kvůli velkým odchylkám ve fenotypové expresi. Každý pacient musí být hodnocen a léčen na základě své specifické prezentace, aby se obnovila estetická a funkční rovnováha.[7]
Chirurgické korekce hlavních příznaků;
- Kraniosynostóza korekce: Preferovaný věk pro tento postup je mezi 6–9 měsíci věku.[17] Provedení této operace v tak raném věku může omezit další vývoj asymetrie obličeje, pokud je asymetrie způsobena kraniosynostózou, a zabrání prodloužené zvýšené nitrolební tlak (ICP).[18] V publikované literatuře však chybí údaje o přesném riziku zvýšeného intrakraniálního tlaku u pacientů s CFND.[7][18] Chirurgický zákrok zahrnuje posun čelní kosti v kombinaci s přestavbou nadočnicového okraje.[19]
- Orbitální hypertelorismus: Je upřednostňováno počkat s touto léčbou až do věku 5–8 let po trvalém chrupu.[7][20] Postupy, které lze provést, jsou: bipartice obličeje a kostní osteotomie. Obličejová bipartice je preferovanou volbou, protože je zapotřebí méně dalších korekcí a také poskytuje stabilnější dlouhodobý výsledek po léčbě.[18] Po korekci orbit jsou mediální rohy očí umístěny více do vodorovné čáry.[7]
- Korekce nosní deformity: Korekce široké nosní základny se provádí současně s opravou orbitálního hypertelorismu. To je pro dobré vyrovnání očí s nosem pro nejlepší estetický výsledek. Bifidní špička nosu bude ošetřena až ve věku 18 let, kdy kostra pacienta plně dospěla.[7][21]
Epidemiologie
Kraniofrontonazální dysplazie je velmi vzácný genetický stav. Ve zveřejněné literatuře je tedy málo informací a neexistuje shoda ohledně epidemiologických statistik.
Hlášené hodnoty výskytu se pohybovaly od 1: 100 000 do 1: 120 000.[3]
Reference
- ^ A b C d E Wieland, I., Jakubiczka, S., Muschke, P. a kol. Mutace genu pro efrin-B1 způsobují kraniofrontonasální syndrom. Am J Hum Genet 74: 1209-1215, 2004.
- ^ A b C d E F Twigg, S. R., Kan, R., Babbs, C. a kol. Mutace efrinu-B1 (EFNB1), markeru tvorby hranice tkáně, způsobují kraniofrontonasální syndrom. Proc Natl Acad Sci USA 101: 8652-8657, 2004.
- ^ A b C d E F G h i j „Archivovaná kopie“ (PDF). Archivovány od originál (PDF) dne 12.9.2014. Citováno 2012-11-03.CS1 maint: archivovaná kopie jako titul (odkaz)
- ^ A b C d E Zafeiriou, D. I., Pavlidou, E. L., Vargiami, E., et al. Různé klinické a genetické aspekty kraniofrontonazálního syndromu. Pediatr Neurol. Únor 2011; 44 (2): 83-7.
- ^ A b C Grutzner, E., Gorlin, R. J. Kraniofrontonazální dysplázie: fenotypová exprese u žen a mužů a genetické úvahy. Oral Surg Oral Med Oral Pathol 65: 436-444, 1988.
- ^ A b C d E F G h Kawamoto, H. K., Heller, J. B., Heller, M. M. a kol. Kraniofrontonazální dysplazie: algoritmus chirurgické léčby. Plast Reconstr Surg 120: 1943-1956, 2007.
- ^ A b Kullander, K., Klein, R. Mechanismy a funkce signalizace Eph a ephrin. Nat Rev Mol Cell Biol 3: 475-86, 2002.
- ^ Wilkinson, D. G. Několik rolí receptorů EPH a efrinů v nervovém vývoji. Nat Rev Neurosci. 2001; 2: 155–164.
- ^ A b Beutler, E., Yeh, M., Fairbanks, V. F. Normální lidská žena jako mozaika aktivity X-chromozomu: studie využívající gen jako marker pro nedostatek G-6-PD. Proc Natl Acad Sci U S A 48: 9–16, 1962.
- ^ A b Wieland, I., Makarov, R., Reardon, W., Tinschert, S., Goldenberg, A., Thierry, P. a kol. Rozdělení molekulárních mechanismů u kraniofrontonasálního syndromu: diferenciální exprese mRNA mutantního EFNB1 a buněčné mozaiky. Eur J Hum Genet. Únor 2008; 16 (2): 184-91.
- ^ Apostolopoulou, D., Stratoudakis, A., Hatzaki, A. Novel de Novo Mutation Within EFNB1 Gene in a Young Girl With Craniofrontonasal Syndrome. Rozštěp paláce Craniofac J. 49: 109-13, 2012.
- ^ A b Wieland, I., Reardon, W., Jakubiczka, S. a kol. Dvacet šest nových mutací EFNB1 u familiárního a sporadického kraniofrontonasálního syndromu (CFNS). Hum Mutat 26: 113-118, 2005.
- ^ A b Twigg, S. R., Matsumoto, K., Kidd, A. M. a kol. Původ mutací EFNB1 v kraniofrontonasálním syndromu: častý somatický mozaicismus a vysvětlení nedostatku nosných mužů. Am J Hum Genet 78: 999-1010, 2006.
- ^ A b Wallis, D., Lacbawan, F., Jain, M. a kol. Další mutace EFNB1 u kraniofrontonazálního syndromu. Am J Med Genet A 146A: 2008-2012, 2008.
- ^ https://www.cdc.gov/mmwr/PDF/rr/rr4409.pdf
- ^ Panchal, J. a kol. Léčba kraniosynostózy. Plast Reconstr Surg. 2003 květen; 111 (6): 2032-48
- ^ A b C van den Elzen, M. E., Wolvius, E. B. a kol. Dlouhodobý chirurgický výsledek pro kraniofaciální deformity pacientů s kraniofrontonasální dysplázií s prokázanými mutacemi EFNB1. J Plast Reconstr. Surg.
- ^ „Archivovaná kopie“ (PDF). Archivovány od originál (PDF) dne 2012-06-17. Citováno 2012-11-03.CS1 maint: archivovaná kopie jako titul (odkaz)
- ^ Bentz, M. L. Dětská plastická chirurgie; Kapitola 9 Hypertelorismus, autor: Renato Ocampo, Jr., MD / John A. Persing, MD
- ^ van den Elzen, M. E., Versnel, S. L. a kol. Dlouhodobé výsledky po 40 letech zkušeností s léčbou vzácných rozštěpů obličeje: Část 2 e symetrické střední rozštěpy. J Plast Reconstr Aesthet Surg 64 (10): 1344-52, 2011.
externí odkazy
- Kraniofrontonazální dysplázie v NIH kancelář Vzácné nemoci
- Kraniofrontonazální dysplazie Typ Teebi v NIH kancelář Vzácné nemoci
| Klasifikace | |
|---|---|
| Externí zdroje |