Dilatační kardiomyopatie - Dilated cardiomyopathy
| Dilatační kardiomyopatie | |
|---|---|
| Ostatní jména | Městnavá kardiomyopatie, idiopatická kardiomyopatie, primární kardiomyopatie[1] |
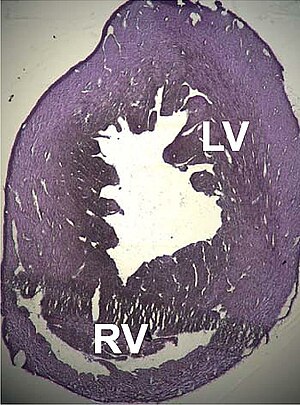 | |
| Myší srdce plátek ukazuje dilatační kardiomyopatie | |
| Specialita | Kardiologie |
| Příznaky | Cítit se unaveně, otoky nohou, dušnost, bolest na hrudi, mdloby[2] |
| Komplikace | Srdeční selhání, onemocnění srdeční chlopně, nepravidelný srdeční tep[3][4] |
| Obvyklý nástup | Střední věk[5] |
| Typy | Tachykardie,[6][7] ostatní |
| Příčiny | Genetika, alkohol, kokain určité toxiny, komplikace těhotenství, jisté infekce[8][9][7] |
| Diagnostická metoda | Podporováno elektrokardiogram, rentgen hrudníku, echokardiogram[9] |
| Diferenciální diagnostika | Ischemická choroba srdeční, onemocnění srdeční chlopně, plicní embolie, jiný kardiomyopatie[5] |
| Léčba | Změny životního stylu, léky, implantovatelný kardioverterový defibrilátor, transplantace srdce[9] |
| Léky | ACE inhibitor, beta blokátor, diuretický, ředidla krve[9] |
| Prognóza | Pětiletá míra přežití ~50%[9] |
| Frekvence | 1 z 2 500[9] |
Dilatační kardiomyopatie (DCM) je stav, ve kterém srdce se zvětší a nemůže pumpovat krev účinně.[3] Příznaky se liší od žádného po pocit únavy, otoky nohou, a dušnost.[2] Může také vést k bolest na hrudi nebo mdloby.[2] Komplikace mohou zahrnovat srdeční selhání, onemocnění srdeční chlopně, nebo nepravidelný srdeční tep.[3][4]
Příčiny zahrnují genetika, alkohol, kokain, některé toxiny, komplikace těhotenství a určité infekce.[8][9] Ischemická choroba srdeční a vysoký krevní tlak mohou hrát roli, ale nejsou primární příčinou.[5][8] V mnoha případech zůstává příčina nejasná.[8] Je to typ kardiomyopatie, skupina nemocí, která primárně postihuje srdeční sval.[3] Diagnóza může být podpořena elektrokardiogram, rentgen hrudníku nebo echokardiogram.[9]
U pacientů se srdečním selháním může léčba zahrnovat léky v ACE inhibitor, beta blokátor, a diuretický rodiny.[9] A strava s nízkým obsahem soli může být také užitečné.[5] U pacientů s určitými typy nepravidelného srdečního rytmu ředidla krve nebo implantovatelný kardioverterový defibrilátor může být doporučeno.[9] Pokud jiná opatření nejsou účinná a transplantace srdce může být v některých možnost.[9]
Postiženo je přibližně 1 na 2 500 lidí.[9] Vyskytuje se častěji u mužů než u žen.[10] Nástup je nejčastěji ve středním věku.[5] Pětiletá míra přežití je asi 50%.[9] Může se také vyskytnout u dětí a je nejběžnějším typem kardiomyopatie v této věkové skupině.[9]
Příznaky a symptomy
Dilatační kardiomyopatie se vyvíjí zákerne a nemusí zpočátku způsobovat dostatečně významné příznaky, které by ji mohly ovlivnit kvalita života.[11][12] Mnoho lidí však má značné příznaky. Mohou zahrnovat:
- Dušnost
- Synkopa (mdloby)
- Angina, ale pouze za přítomnosti ischemická choroba srdeční
Osoba trpící dilatační kardiomyopatií může mít zvětšené srdce, s plicní otok a zvýšené krční žilní tlak a nízká pulzní tlak. Známky mitrální a trikuspidální regurgitace může být přítomen.[12]
Příčiny
Ačkoli v mnoha případech není zřejmá žádná příčina, dilatační kardiomyopatie je pravděpodobně výsledkem poškození myokard vyráběné různými toxický, metabolické nebo infekční agens. Může to být způsobeno vláknitou změnou myokardu oproti předchozímu infarkt myokardu. Nebo to mohou být pozdní následky akutního viru myokarditida, například s Virus Coxsackie B. a další enteroviry[13] pravděpodobně zprostředkováno prostřednictvím imunologického mechanismu.[14]
Mezi další příčiny patří:
- Chagasova choroba, kvůli Trypanosoma cruzi. Toto je nejčastější infekční příčina dilatační kardiomyopatie v Latinské Americe[15]
- Těhotenství. Dilatační kardiomyopatie se vyskytuje pozdě v těhotenství nebo několik týdnů až měsíců po porodu jako a peripartální kardiomyopatie.[13] V polovině případů je reverzibilní.[13]
- Alkohol zneužívání (alkoholická kardiomyopatie )[13]
- Nealkoholické toxické urážky zahrnují podávání určitých chemoterapeutická činidla, zejména doxorubicin (Adriamycin) a kobalt.[13]
- Nemoc štítné žlázy[12]
- Zánětlivá onemocnění, jako je sarkoidóza a pojivové tkáně[12]
- Kardiomyopatie vyvolaná tachykardií[7]
- Svalová dystrofie
- Tuberkulóza - 1 až 2% případů TB.[16]
- Autoimunitní mechanismy[17]
- Nedostatek thiaminu
Nedávné studie ukázaly, že u subjektů s extrémně vysokým výskytem (několik tisíc denně) předčasné komorové kontrakce (extrasystola) může vyvinout dilatační kardiomyopatii. V těchto případech, pokud je extrasystola redukována nebo odstraněna (například pomocí ablační terapie), kardiomyopatie obvykle ustupuje.[18][19]
Genetika
| Genetické asociace s dilatační kardiomyopatií | |||
|---|---|---|---|
| Typ | OMIM | Gen | Místo |
| CMD1A | 115200 | LMNA | 1q21 |
| CMD1B | 600884 | neznámý (TMOD1 kandidát) | 9q13 |
| CMD1C | 601493 | LDB3 | 10q22-q23 |
| CMD1D | 601494 | TNNT2 | 1q32 |
| CMD1E | 601154 | SCN5A | 3p |
| CMD1F | 602067 | 6q23 | |
| CMD1G | 604145 | TTN | 2q31 |
| CMD1H | 604288 | 2q14-q22 | |
| CMD1I | 604765 | DES | |
| CMD1K | 605582 | 6q12-q16 | |
| CMD1L | 606685 | SGCD | 5q33 |
| CMD1M | 607482 | CSRP3 | 11p15.1 |
| CMD1N | 607487 | TCAP | 17q12 |
| CMD1O | 608569 | ABCC9 | 12p12.1 |
| CMD1P | 609909 | PLN | 6q22.1 |
| CMD1Q | 609915 | 7q22.3-q31.1 | |
| CMD1R | ACTC | 15q14 | |
| CMD1S | MYH7 | 14q12 | |
| CMD1T | TMPO | 12q22 | |
| CMD1U | PSEN1 | 14q24.3 | |
| CMD1V | PSEN2 | 1q31-q42 | |
| CMD1W | 611407 | VCL | 10q22-q23 |
| CMD1X | FCMD | 9q31 | |
| CMD1Y | 611878 | TPM1 | 15q22.1 |
| CMD1Z | 611879 | TNNC1 | 3p21.3-p14.3 |
| CMD1AA | 612158 | ACTN2 | 1q42-q43 |
| CMD2A | 611880 | TNNI3 | 19q13.4 |
| CMD3A | 300069 | TAZ | Xq28 |
| CMD3B | 302045 | DMD | Xp21.2 |
| ALPK3 | 15q25.3 | ||
Asi 25–35% postižených jedinců má familiární formy onemocnění,[13] s většinou mutace ovlivňující kódování genů cytoskeletální bílkoviny,[13] zatímco některé ovlivňují jiné proteiny podílející se na kontrakci.[20] Nemoc je geneticky heterogenní, ale nejběžnější formou přenosu je autosomálně dominantní vzor.[13]Autosomálně recesivní (jak je uvedeno například v Alströmův syndrom[13]), X-vázaný (jako v Duchennova svalová dystrofie ), a mitochondriální zjistí se také dědičnost nemoci.[21] Někteří příbuzní osob postižených dilatační kardiomyopatií mají předklinické, asymptomatické změny srdečního svalu.[22]
Mezi další cytoskeletální proteiny zapojené do DCM patří α-srdeční aktin, desmin a nukleární lamináty A a C.[13] Mitochondriální delece a mutace pravděpodobně způsobují DCM změnou myokardu Generování ATP.[13]
Kayvanpour a kol. 2016 provedlo metaanalýzu s největším souborem dostupných dat o asociacích genotyp-fenotyp v DCM a mutacích v lamin (LMNA), fosfolambanu (PLN), RNA Binding Motif Protein 20 (RBM20), Cardiac Myosin Binding Protein C (MYBPC3), Myosin Těžký řetězec 7 (MYH7), srdeční troponin T 2 (TNNT2) a srdeční troponin I (TNNI3). Rovněž přezkoumali nedávné studie zkoumající asociace genotyp-fenotyp u pacientů s DCM s mutacemi titinu (TTN). Nosiči mutací LMNA a PLN vykazovali vysokou prevalenci srdeční transplantace a ventrikulární arytmie. Ukázalo se, že dysrytmie a náhlá srdeční smrt (SCD) se vyskytují ještě před projevy DCM a symptomů srdečního selhání u nosičů mutací LMNA.[23]
Patofyziologie
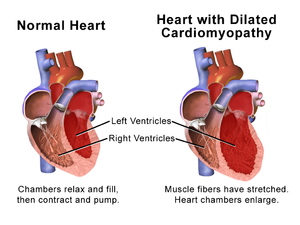
Progrese srdečního selhání je spojena s remodelací levé komory, která se projevuje postupným zvyšováním koncových diastolických a koncových objemů levé komory, ztenčováním stěn a změnou geometrie komory na sférickější a méně protáhlý tvar. Tento proces je obvykle spojen s trvalým poklesem ejekční frakce. Koncept remodelace srdce byl původně vyvinut k popisu změn, ke kterým dochází ve dnech a měsících po infarktu myokardu.[24]
Kompenzační účinky
Jak DCM postupuje, aktivují se dva kompenzační mechanismy v reakci na zhoršenou kontraktilitu myocytů a snížený zdvihový objem:[12]
- Frank-Starlingův zákon
- Neurohormonální zpětná vazba prostřednictvím aktivace podpůrný nervový systém a renin-angiotensin Systém.
Tyto reakce zpočátku kompenzují snížený srdeční výdej a udržují odpovědi s DCM asymptomatické. Nakonec se však tyto mechanismy stanou škodlivými, intravaskulární objem se stane příliš velkým a progresivní dilatace vede k příznakům srdečního selhání.
Výpočtové modely
Srdeční dilatace je příčná izotropní, nevratný proces vyplývající z přebytečných kmenů na myokard.[25] Výpočtový model objemového, izotropního a růstu srdeční stěny předpovídá vztah mezi srdečními kmeny (např. Objemovým přetížením po infarktu myokardu) a dilatací pomocí následujících řídících rovnic:

kde je elastický objemový úsek, který je reverzibilní a je nevratný, izotropní růst objemu popsaný:


![F^{g}={mathbb {I}}+[lambda ^{{g}}-1]f_{{0}}otimes f_{{0}},](https://wikimedia.org/api/rest_v1/media/math/render/svg/ad6fd285b02f97569017cca85b9c109c147fbfbe)
kde je vektor, který ukazuje podél a kardiomyocyt je dlouhá osa a je úsek kardiomyocytů v důsledku růstu. Celkový růst kardiomyocytů je dán vztahem:



Výše uvedený model odhaluje postupné rozšiřování myokard, zejména komorový myokard, pro podporu krve objemové přetížení v komorách. Dilatace se projevuje zvýšením celkové srdeční hmoty a srdečního průměru. Kardiomyocyty dosahují maximální délky 150 mv endokardu a 130 mv epikardu přidáním sarkomér.[26] Kvůli zvětšení průměru vypadá rozšířené srdce sférického tvaru, na rozdíl od eliptického tvaru zdravého lidského srdce. Kromě toho si stěny komor zachovávají stejnou tloušťku, která je charakteristická pro patofyziologickou dilataci srdce.

Chlopňové účinky
Jak se komory zvětšují, mitrální i trikuspidální chlopně mohou ztratit schopnost správně se spojit. Tato ztráta spolupráce může vést k mitrální a trikuspidální regurgitace. Ve výsledku je u pacientů s DCM zvýšené riziko fibrilace síní. Dále se zmenší objem mrtvice a na komoru se umístí větší objemová zátěž, čímž se zvýší příznaky srdečního selhání.[12]
Diagnóza


Generalizované zvětšení srdce je vidět na normálu rentgen hrudníku. Pleurální výpotek může být také zaznamenáno, což je způsobeno plicní žilní hypertenzí.
The elektrokardiogram často ukazuje sinusová tachykardie nebo fibrilace síní, ventrikulární arytmie, zvětšení levé síně, a někdy defekty intraventrikulárního vedení a nízké napětí. Když blok levé větve svazku (LBBB) doprovází odchylka pravé osy (RAD) je vzácná kombinace považována za vysoce sugestivní pro dilatační nebo městnavou kardiomyopatii.[27][28] Echokardiogram ukazuje dilataci levé komory s normálními nebo ztenčenými stěnami a redukovanou ejekční frakce. Srdeční katetrizace a koronární angiografie jsou často prováděny k vyloučení ischemické choroby srdeční.
Genetické testování může být důležité, protože jedna studie ukázala, že genové mutace v genu TTN (který kóduje protein zvaný titin ) jsou odpovědní za „přibližně 25% familiárních případů idiopatické dilatační kardiomyopatie a 18% sporadických případů“.[29] Výsledky genetického testování mohou lékařům a pacientům pomoci pochopit základní příčinu dilatační kardiomyopatie. Výsledky genetických testů mohou také pomoci při rozhodování o tom, zda by příbuzní pacienta měli podstoupit genetické testování (aby se zjistilo, zda mají stejnou genetickou mutaci) a vyšetření srdce, aby se prověřil časný nález rozšířené kardiomyopatie.
Srdeční magnetická rezonance (srdeční MRI) může také poskytnout užitečné diagnostické informace u pacientů s dilatační kardiomyopatií.[30]
Léčba
Lékařská terapie
Léková terapie může zpomalit progresi a v některých případech dokonce zlepšit stav srdce. Standardní terapie může zahrnovat omezení solí, ACE inhibitory, diuretika, a beta-blokátory.[12] Antikoagulancia lze také použít pro antitrombotickou terapii. Existují určité důkazy o výhodách koenzym Q10 při léčbě srdečního selhání.[31][32][33]
Elektrické ošetření
Umělé kardiostimulátory mohou být použity u pacientů se zpožděním intraventrikulárního vedení a implantovatelné kardioverter-defibrilátory u pacientů s rizikem arytmie. Ukázalo se, že tyto formy léčby zabraňují náhlé srdeční smrti, zlepšují příznaky a snižují hospitalizaci u pacientů se systolickým srdečním selháním.[34]
Chirurgická léčba
U pacientů s pokročilým onemocněním, kteří neodolávají lékařské terapii, transplantace srdce lze zvážit. U těchto lidí se roční přežití blíží 90% a více než 50% přežívá déle než 20 let.[34]
Epidemiologie
Ačkoli je toto onemocnění častější u afroameričanů než u bělochů,[35] může se vyskytnout u jakékoli populace pacientů.
Pokyny k výzkumu
Byly zkoumány terapie podporující reverzní remodelaci, což může naznačovat nový přístup k prognóze kardiomyopatií (viz remodelace komor ).[24][36]
Ostatní zvířata
Dilatovaná kardiomyopatie je dědičné onemocnění u některých plemen psů, včetně Boxer, Dobrman, německá doga, Irský vlkodav, a St Bernard.[37] Léčba je založena na léčbě, včetně ACE inhibitorů, kličková diuretika, a inhibitory fosfodiesterázy. V roce 2019 vědci z University of California, Davis School of Veterinary Medicine publikoval a zpráva popisující souvislost mezi určitými dietami a rozvojem dilatační kardiomyopatie u plemen psů postrádajících genetickou predispozici, zejména u Zlatí retrívři. Diety spojené s DCM byly popsány jako „BEG“ (butik, exotická přísada a / nebo bez obilovin) krmiva pro psy, stejně jako diety bohaté na luštěniny. Pro léčbu DCM souvisejícího se stravou jsou obvykle indikovány změny v potravinách a suplementace taurinem, podle potřeby spolu s tradičními způsoby léčby.
Dilatovaná kardiomyopatie je také onemocnění postihující některá plemena koček, včetně Orientální krátkosrstá, Barmská, Peršan, a Habešský. U koček je nedostatek taurinu nejčastější příčinou dilatační kardiomyopatie.[38] Na rozdíl od těchto dědičných forem byl v celkové populaci koček před přidáním taurinu do komerčního krmiva pro kočky běžný nedědičný DCM.
Existuje také vysoký výskyt dědičné rozšířené kardiomyopatie v zajetí Zlatí křečci (Mesocricetus auratus), v nemalé míře za to, že jsou vysoce inbrední. Výskyt je dostatečně vysoký na to, aby bylo vyvinuto několik kmenů zlatého křečka, které slouží jako zvířecí modely při klinickém testování lidských forem onemocnění.[39]
Reference
- ^ „Jiná jména pro kardiomyopatii“. NHLBI. 22. června 2016. Citováno 31. srpna 2016.
- ^ A b C „Jaké jsou příznaky a příznaky kardiomyopatie?“. NHLBI. 22. června 2016. Citováno 10. listopadu 2017.
- ^ A b C d „Co je to kardiomyopatie?“. NHLBI. 22. června 2016. Citováno 10. listopadu 2017.
- ^ A b „Druhy kardiomyopatie“. NHLBI. 22. června 2016. Citováno 10. listopadu 2017.
- ^ A b C d E Ferri, Fred F. (2017). E-kniha Ferriho klinického poradce 2018: 5 knih v 1. Elsevier Health Sciences. str. 244. ISBN 9780323529570.
- ^ „Kardiomyopatie vyvolaná tachykardií“. Evropská kardiologická společnost. 2019-03-29. Citováno 2019-03-29.
Kardiomyopatie vyvolaná tachykardií je reverzibilní příčinou srdečního selhání a dilatační kardiomyopatie. Kardiomyopatie vyvolaná tachykardií by měla být zvážena u všech pacientů s dilatační kardiomyopatií nejistého původu, kteří mají tachykardii nebo fibrilaci síní s rychlou komorovou frekvencí.
- ^ A b C Umana, Ernesto; Solares, C. Arturo; Alpert, Martin A (01.01.2003). "Kardiomyopatie vyvolaná tachykardií". American Journal of Medicine. 114 (1): 51–55. doi:10.1016 / S0002-9343 (02) 01472-9. PMID 12543289.
- ^ A b C d „Co způsobuje kardiomyopatii?“. NHLBI. 22. června 2016. Citováno 10. listopadu 2017.
- ^ A b C d E F G h i j k l m n Weintraub, RG; Semsarian, C; Macdonald, P (22. července 2017). „Dilatační kardiomyopatie“. Lanceta. 390 (10092): 400–414. doi:10.1016 / S0140-6736 (16) 31713-5. PMID 28190577. S2CID 46801202.
- ^ „Kdo je ohrožen kardiomyopatií? - NHLBI, NIH“. NHLBI. 22. června 2016. Citováno 10. listopadu 2017.
- ^ Watkins, Hugh; Ashrafian, Houman; Redwood, Charles (2011-04-27). „Zděděné kardiomyopatie“. New England Journal of Medicine. 364 (17): 1643–1656. doi:10.1056 / nejmra0902923. PMID 21524215.
- ^ A b C d E F G Patofyziologie srdečních chorob: společný projekt studentů medicíny a fakulty. Lilly, Leonard S., Harvard Medical School. (5. vydání). Baltimore, MD: Wolters Kluwer / Lippincott Williams & Wilkins. 2011. ISBN 9781605477237. OCLC 649701807.CS1 maint: ostatní (odkaz)
- ^ A b C d E F G h i j k Mitchell, Richard Sheppard; Kumar, Vinay; Abbas, Abul K .; Fausto, Nelson (2007). Robbinsova základní patologie (8. vydání). Philadelphia: Saunders. ISBN 978-1-4160-2973-1.
- ^ Martino TA, Liu P, Sole MJ (únor 1994). „Virová infekce a patogeneze rozšířené kardiomyopatie“. Circ. Res. 74 (2): 182–8. doi:10.1161 / 01.res.74.2.182. PMID 8293557.
- ^ „Dilatovaná kardiomyopatie - kardiovaskulární poruchy“.
- ^ Agarwal Ritesh, Malhotra Puneet, Awasthi Anshu, Kakkar Nandita, Gupta Dheeraj (2005). „Tuberkulózní dilatační kardiomyopatie: nedostatečně uznávaná entita?“. BMC Infect Dis. 5: 29. doi:10.1186/1471-2334-5-29. PMC 1090580. PMID 15857515.CS1 maint: více jmen: seznam autorů (odkaz)
- ^ San Martín MA, García A, Rodríguez FJ, Terol I (květen 2002). „[Dilatační kardiomyopatie a autoimunita: přehled současných znalostí a perspektiv]“. Rev Esp Cardiol. (ve španělštině). 55 (5): 514–24. doi:10.1016 / s0300-8932 (02) 76644-x. PMID 12015932. Archivovány od originál dne 09.01.2009.
- ^ Belhassen B (duben 2005). „Radiofrekvenční ablace„ benigních “extrasystolů výtokového traktu pravé komory: terapie, která zjistila své onemocnění?“. J. Am. Sb. Cardiol. 45 (8): 1266–8. doi:10.1016 / j.jacc.2005.01.028. PMID 15837260.
- ^ Shiraishi H, Ishibashi K, Urao N a kol. (Listopad 2002). „Případ kardiomyopatie vyvolané předčasnými komorovými komplexy“. Circ. J. 66 (11): 1065–7. doi:10.1253 / cirk.66.1065. PMID 12419942.
- ^ Ross J (březen 2002). „Dilatovaná kardiomyopatie: koncepty odvozené z genově deficitních a transgenních zvířecích modelů“. Circ. J. 66 (3): 219–24. doi:10.1253 / cirk.66.219. PMID 11922267.
- ^ Schönberger J, Seidman CE (srpen 2001). „Mnoho cest vede ke zlomenému srdci: genetika dilatační kardiomyopatie“. American Journal of Human Genetics. 69 (2): 249–60. doi:10.1086/321978. PMC 1235300. PMID 11443548.
- ^ Mahon NG, Murphy RT, MacRae CA, Caforio AL, Elliott PM, McKenna WJ (červenec 2005). „Echokardiografické hodnocení u asymptomatických příbuzných pacientů s dilatační kardiomyopatií odhaluje preklinické onemocnění“. Annals of Internal Medicine. 143 (2): 108–15. doi:10.7326/0003-4819-143-2-200507190-00009. PMID 16027452. S2CID 22278646.
- ^ Kayvanpour, Elham; Sedaghat-Hamedani, Farbod; Amr, Ali; Lai, Alan; Haas, Jaan; Holzer, Daniel B .; Frese, Karen S .; Keller, Andreas; Jensen, Katrin; Katus, Hugo A .; Meder, Benjamin (2016-08-30). „Sdružení genotyp-fenotyp v dilatované kardiomyopatii: metaanalýza u více než 8 000 jedinců“. Klinický výzkum v kardiologii. 106 (2): 127–139. doi:10.1007 / s00392-016-1033-6. PMID 27576561. S2CID 27511518.
- ^ A b Pieske B (2004). „Reverzní přestavba srdečního selhání - skutečnost nebo fikce?“. Eur Heart J Suppl. 6: D66–78. doi:10.1016 / j.ehjsup.2004.05.019.
- ^ Goektepe, Serdar; Abilez, Oscar John; Kuhl, Ellen (2010). "Obecný přístup k omezenému růstu s příklady srdce sportovce, dilatace srdce a zesílení srdeční stěny". Mechanika a fyzika pevných látek. 58 (10): 1661–1680. Bibcode:2010JMPSo..58.1661G. doi:10.1016 / j.jmps.2010.07.003.
- ^ Goektepe, Serdar; Abilez, Oscar John; Parker, K; Kuhl, Ellen (2010). "Víceúrovňový model pro excentrický a koncentrický růst srdce prostřednictvím sarkomerogeneze". Teoretická biologie. 58: 1661–1680. Bibcode:2010JMPSo..58.1661G. doi:10.1016 / j.jmps.2010.07.003.
- ^ Nikolic G, Marriott HJ (říjen 1985). "Blok větve levého svazku s odchylkou pravé osy: značka kongestivní kardiomyopatie". J Elektrokardiol. 18 (4): 395–404. doi:10.1016 / s0022-0736 (85) 80022-4. PMID 3906012.
- ^ Childers R, Lupovich S, Sochanski M, Konarzewska H (2000). "Odbočný blok levého svazku a odchylka pravé osy: zpráva o 36 případech". J Elektrokardiol. 33 (Suppl): 93–102. doi:10.1054 / jclc.2000.20326. PMID 11265743.
- ^ Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE (16. února 2012). „Zkrácení titinu způsobující dilatační kardiomyopatii“. N Engl J Med. 366 (7): 619–628. doi:10.1056 / NEJMoa1110186. PMC 3660031. PMID 22335739.
- ^ Pennell DJ, Sechtem UP, Higgins CB, Manning WJ, Pohost GM, Rademakers FE, van Rossum AC, Shaw LJ, Yucel EK (listopad 2004). „Klinické indikace pro kardiovaskulární magnetickou rezonanci (CMR): zpráva konsensuálního panelu“. Eur Heart J. 25 (21): 1940–1965. doi:10.1016 / j.ehj.2004.06.040. PMID 15522474.
- ^ Langsjoen PH, Langsjoen PH, Folkers K (1990). „Šestiletá klinická studie léčby kardiomyopatie koenzymem Q10“. Int J Tissue React. 12 (3): 169–71. PMID 2276895.
- ^ Folkers K, Langsjoen P, Langsjoen PH (leden 1992). „Terapie koenzymem Q10 u pacientů se srdečním selháním, kteří jsou způsobilí nebo nezpůsobilí k transplantaci“. Biochem Biophys Res Commun. 182 (1): 247–53. doi:10.1016 / S0006-291X (05) 80137-8. PMID 1731784.
- ^ Baggio E, Gandini R, Plancher AC, Passeri M, Carmosino G (1994). „Italská multicentrická studie o bezpečnosti a účinnosti koenzymu Q10 jako doplňkové léčby srdečního selhání. Vyšetřovatelé drogového dozoru CoQ10“. Mol. Aspects Med. 15 (Suppl): s287–94. doi:10.1016 / 0098-2997 (94) 90040-X. PMID 7752841.
- ^ A b McPhee, Stephen J .; Rabow, Michael W .; Papadakis, Maxine A. (01.09.2016). Aktuální lékařská diagnóza a léčba 2017. Papadakis, Maxine A. ,, McPhee, Stephen J. ,, Rabow, Michael W. (padesáté šesté vydání). New York. ISBN 978-1259585111. OCLC 957316517.
- ^ Coughlin SS, Labenberg JR, Tefft MC (březen 1993). „Černobílé rozdíly v idiopatické dilatační kardiomyopatii: Washington DC DC dilatační studie kardiomyopatie“. Epidemiologie. 4 (2): 165–72. doi:10.1097/00001648-199303000-00013. PMID 8452906.
- ^ Reis Filho JR, Cardoso JN, Cardoso CM, Pereira-Barretto AC (2015). „Reverzní přestavba srdce: značka lepší prognózy při srdečním selhání“. Arq. Podprsenky. Cardiol. 104 (6): 502–6. doi:10.5935 / abc.20150025. PMC 4484683. PMID 26131706.
- ^ Oyama MA, Chittur S (červenec 2005). "Genomické expresní vzorce srdečních tkání u psů s dilatační kardiomyopatií". Am J Vet Res. 66 (7): 1140–55. doi:10.2460 / ajvr.2005.66.1140. PMID 16111151.
- ^ Pion, P. D .; Kittleson, M. D .; Thomas, W. P .; Skiles, M. L .; Rogers, Q. R. (1992-07-15). „Klinické nálezy u koček s dilatační kardiomyopatií a vztah nálezů k nedostatku taurinu“. Journal of the American Veterinary Medical Association. 201 (2): 267–274. ISSN 0003-1488. PMID 1500323.
- ^ Nigro V, Okazaki Y, Belsito A a kol. (Duben 1997). „Identifikace genu kardiomyopatie křečka syrského“. Hučení. Mol. Genet. 6 (4): 601–7. doi:10,1093 / hmg / 6,4601. PMID 9097966.
externí odkazy
- Dilatační kardiomyopatie informace pro rodiče.
| Klasifikace | |
|---|---|
| Externí zdroje |