Dědičná spastická paraplegie - Hereditary spastic paraplegia
| Dědičná spastická paraplegie | |
|---|---|
| Specialita | Neurologie |
Dědičná spastická paraplegie (HSP) je skupina dědičných onemocnění, jejichž hlavním rysem je progresivní porucha chůze. Onemocnění má progresivní tuhost (spasticita ) a kontrakce na dolních končetinách.[1] HSP je také známá jako dědičná spastická paraparéza, familiární spastická paraplegie, francouzská choroba osídlení, Strumpellova choroba nebo Strumpell-Lorrainova choroba. Příznaky jsou výsledkem dlouhodobé dysfunkce axony v mícha. Ovlivněné buňky jsou primární motorické neurony; nemoc je tedy horní motorický neuron choroba.[2] HSP není forma dětská mozková obrna i když se to fyzicky může zdát a chovat se stejně spastická diplegie. Původ HSP se liší od mozkové obrny. Navzdory tomu se některé stejné léky proti spasticitě používají v spastická mozková obrna se někdy používají k léčbě příznaků HSP.
HSP je způsoben vadami v transportu proteinů, strukturálních proteinů, proteinů udržujících buňky, lipidů a dalších látek skrz buňku. Jsou ovlivněna dlouhá nervová vlákna (axony), protože díky dlouhé vzdálenosti jsou nervové buňky zvláště citlivé na defekty těchto zmíněných mechanismů.[3][4]
Nemoc byla poprvé popsána v roce 1880 německým neurologem Adolph Strümpell.[5] Podrobněji to popsal v roce 1888 francouzský lékař Maurice Lorrain.[6] Vzhledem k jejich příspěvku při popisu nemoci se ve frankofonních zemích stále nazývá Strümpell-Lorrainova choroba. Termín dědičná spastická paraplegie byl vytvořen Anita Hardingová v roce 1983.[7]
Příznaky a symptomy
Příznaky závisí na typu zděděného HSP. Hlavní rys nemoci je progresivní spasticita v dolních končetinách kvůli pyramidový trakt dysfunkce. To také vede k rychlým reflexům, extenzorovým plantárním reflexům, svalové slabosti a variabilním poruchám močového měchýře. Kromě toho mezi hlavní příznaky HSP patří také abnormální chůze a potíže s chůzí, snížené vibrační vnímání kotníků a parestézie.[8]Jedinci s HSP mohou pociťovat extrémní únavu spojenou s centrálním nervovým systémem a neuromuskulárními poruchami, které mohou být invalidizující.[9][10][11] Počáteční příznaky jsou obvykle potíže s rovnováhou, bodnutí prstu nebo zakopnutí. Příznaky HSP mohou začít v jakémkoli věku, od kojeneckého věku po starší než 60 let. Pokud příznaky začnou během dospívání nebo později, potom spastická porucha chůze obvykle prochází po mnoho let. Nakonec mohou být vyžadovány hole, chodítka a invalidní vozíky, i když někteří lidé nikdy nevyžadují asistenční zařízení.[12] Bylo popsáno, že postižení postupuje rychleji ve formách pro dospělé.[13]
Přesněji řečeno, pacienti s autosomálně dominantní čistou formou HSP odhalují normální pohyb obličeje a extraokulární oblast. Ačkoli trhnutí čelisti u starších pacientů může být svěží, nedochází k poruchám řeči ani obtížím s polykáním. Svalový tonus a síla horní končetiny jsou normální. Na dolních končetinách je svalový tonus zvýšen na hamstringech, kvadricepsech a kotnících. Slabost je nejpozoruhodnější u iliopsoas, tibialis anterior, a v menší míře, ochromit svaly.[13]V komplexní formě poruchy jsou přítomny další příznaky. Patří mezi ně: periferní neuropatie, amyotrofie, ataxie, mentální postižení, ichtyóza, epilepsie, optický neuropatie, demence, hluchota nebo problémy s řečí, polykáním nebo dýcháním.[14]
Anita Hardingová[7] klasifikoval HSP v čisté a komplikované formě. Čistý HSP představuje spasticita v dolních končetinách, spojené s neurogenní porucha močového měchýře stejně jako nedostatek citlivosti na vibrace (palhypestézie ). Na druhou stranu je HSP klasifikován jako komplexní, když je spasticita dolních končetin kombinována s jakýmkoli dalším neurologickým příznakem.
Tato klasifikace je subjektivní a pacienti s komplexními HSP jsou někdy diagnostikováni s cerebelární ataxií se spasticitou, mentální retardací (se spasticitou) nebo leukodystrofie.[7] Některé z genů uvedených níže byly dříve popsány u jiných onemocnění než HSP. Některé klíčové geny se proto překrývají s jinými skupinami chorob.
Věk nástupu
V minulosti byl HSP klasifikován jako časný nástup začínající v raném dětství nebo pozdější nástup v dospělosti. Věk nástupců má maximálně dva body ve věku 2 a kolem 40 let.[15] Nové poznatky naznačují, že dřívější nástup vede k delšímu trvání nemoci bez ztráty pohyblivosti nebo nutnosti použití invalidního vozíku.[15] To bylo také popsáno dříve, že pozdější nástupní formy se vyvíjejí rychleji.[13]
Způsobit
HSP je skupina genetických poruch. Z toho vyplývá obecně dědictví pravidla a lze je zdědit v autosomálně dominantní, autozomálně recesivní nebo X-vázaný recesivní způsob. Zapojený způsob dědičnosti má přímý dopad na šance na zdědění poruchy. Bylo popsáno více než 70 genotypů a více než 50 genetických lokusů bylo spojeno s tímto stavem.[16] Bylo identifikováno deset genů s autozomálně dominantní dědičností. Jeden z nich, SPG4, představuje ~ 50% všech geneticky vyřešených případů nebo přibližně 25% všech případů HSP.[15] Je známo, že se dvanáct genů dědí autozomálně recesivně. Dohromady tato druhá skupina představuje ~ 1/3 případů.[Citace je zapotřebí ]
Většina pozměněných genů funkci zná, ale u některých zatím nebyla identifikována. Všechny jsou uvedeny v seznamu genů níže, včetně způsobu jejich dědičnosti. Některé příklady jsou spastin (SPG4) a paraplegin (SPG7) jsou obě AAA ATPázy.[17]
Genotypy
Geny jsou označeny SPG (Spastický gen chůze). Lokace genů jsou ve formátu: chromozom - rameno (krátké nebo p: dlouhý nebo q) - číslo pásma. Tato označení jsou pouze pro lidské geny. Lokality se mohou (a pravděpodobně i budou) u jiných organismů lišit. Navzdory množství genů, o nichž je známo, že se na tomto stavu podílejí, ~ u 40% případů musí být identifikována jejich příčina.[18] V tabulce níže SPG? se používá k označení genu, který je spojován s HSP, ale dosud neobdržel oficiální označení genu HSP.
| Genotyp | OMIM | Genový symbol | Genový lokus | Dědictví | Věk nástupu | Jiná jména a vlastnosti |
|---|---|---|---|---|---|---|
| SPG1 | 303350 | L1CAM | Xq28 | X-vázaný recesivní | Brzy | Syndrom MASA |
| SPG2 | 312920 | PLP1 | Xq22.2 | X-vázaný recesivní | Variabilní | Pelizaeus – Merzbacherova choroba |
| SPG3A | 182600 | ATL1 | 14q22.1 | Autozomálně dominantní | Brzy | Strumpellova choroba (tato Wiki) |
| SPG4 | 182601 | SPAST | 2p22.3 | Autozomálně dominantní | Variabilní | |
| SPG5A | 270800 | CYP7B1 | 8q12.3 | Autosomálně recesivní | Variabilní | |
| SPG6 | 600363 | NIPA1 | 15q11.2 | Autozomálně dominantní | Variabilní | |
| SPG7 | 607259 | SPG7 | 16q24.3 | Autozomálně dominantní | Variabilní | |
| SPG8 | 603563 | KIAA0196 | 8q24.13 | Autozomálně dominantní | Dospělý | |
| SPG9A | 601162 | ALDH18A1 | 10q24.1 | Autozomálně dominantní | Dospívající | Katarakta s motorickou neuronopatií, nízkým vzrůstem a abnormalitami skeletu |
| SPG9B | 616586 | ALDH18A1 | 10q24.1 | Autosomálně recesivní | Brzy | |
| SPG10 | 604187 | KIF5A | 13q13.3 | Autozomálně dominantní | Brzy | |
| SPG11 | 604360 | SPG11 | 15q21.1 | Autosomálně recesivní | Variabilní | |
| SPG12 | 604805 | RTN2 | 19q13.32 | Autozomálně dominantní | Brzy | |
| SPG13 | 605280 | HSP60 | 2q33.1 | Autozomálně dominantní | Variabilní | |
| SPG14 | 605229 | ? | 3q27 – q28 | Autosomálně recesivní | Dospělý | |
| SPG15 | 270700 | ZFYVE26 | 14q24.1 | Autosomálně recesivní | Brzy | |
| SPG16 | 300266 | ? | Xq11.2 | X-vázaný recesivní | Brzy | |
| SPG17 | 270685 | BSCL2 | 11q12.3 | Autozomálně dominantní | Dospívající | |
| SPG18 | 611225 | ERLIN2 | 8p11.23 | Autosomálně recesivní | Brzy | |
| SPG19 | 607152 | ? | 9q | Autozomálně dominantní | Dospělý nástup | |
| SPG20 | 275900 | SPG20 | 13q13.3 | Autosomálně recesivní | Časný nástup | Troyerův syndrom |
| SPG21 | 248900 | ACP33 | 15q22.31 | Autosomálně recesivní | Časný nástup | MAST syndrom |
| SPG22 | 300523 | SLC16A2 | Xq13.2 | X-vázaný recesivní | Časný nástup | Allan – Herndon – Dudleyův syndrom |
| SPG23 | 270750 | RIPK5 | 1q32.1 | Autosomálně recesivní | Časný nástup | Lisonův syndrom |
| SPG24 | 607584 | ? | 13q14 | Autosomálně recesivní | Časný nástup | |
| SPG25 | 608220 | ? | 6q23 – q24.1 | Autosomálně recesivní | Dospělý | |
| SPG26 | 609195 | B4GALNT1 | 13q13.3 | Autosomálně recesivní | Časný nástup | |
| SPG27 | 609041 | ? | 10q22.1 – q24.1 | Autosomálně recesivní | Variabilní | |
| SPG28 | 609340 | DDHD1 | 14q22.1 | Autosomálně recesivní | Časný nástup | |
| SPG29 | 609727 | ? | 1p31.1 – p21.1 | Autozomálně dominantní | Dospívající | |
| SPG30 | 610357 | KIF1A | 2q37.3 | Autosomálně recesivní | Dospívající | |
| SPG31 | 610250 | REEP1 | 2p11.2 | Autozomálně dominantní | Časný nástup | |
| SPG32 | 611252 | ? | 14q12 – q21 | Autosomálně recesivní | Dětství | |
| SPG33 | 610244 | ZFYVE27 | 10q24.2 | Autozomálně dominantní | Dospělý | |
| SPG34 | 300750 | ? | Xq24 – q25 | X-vázaný recesivní | Dospívající / dospělý | |
| SPG35 | 612319 | FA2H | 16q23.1 | Autosomálně recesivní | Dětství | |
| SPG36 | 613096 | ? | 12q23 – q24 | Autozomálně dominantní | Dospívající / dospělý | |
| SPG37 | 611945 | ? | 8p21.1 – q13.3 | Autozomálně dominantní | Variabilní | |
| SPG38 | 612335 | ? | 4p16 – p15 | Autozomálně dominantní | Dospívající / dospělý | |
| SPG39 | 612020 | PNPLA6 | 19p13.2 | Autosomálně recesivní | Dětství | |
| SPG41 | 613364 | ? | 11p14.1 – p11.2 | Autozomálně dominantní | Dospívání | |
| SPG42 | 612539 | SLC33A1 | 3q25.31 | Autozomálně dominantní | Variabilní | |
| SPG43 | 615043 | C19orf12 | 19q12 | Autosomálně recesivní | Dětství | |
| SPG44 | 613206 | GJC2 | 1q42.13 | Autosomálně recesivní | Dětství / dospívání | |
| SPG45 | 613162 | NT5C2 | 10q24,32 - q24,33 | Autosomálně recesivní | Dětství | |
| SPG46 | 614409 | GBA2 | 9p13.3 | Autosomálně recesivní | Variabilní | |
| SPG47 | 614066 | AP4B1 | 1p13.2 | Autosomálně recesivní | Dětství | |
| SPG48 | 613647 | AP5Z1 | 7p22.1 | Autosomálně recesivní | 6. dekáda | |
| SPG49 | 615041 | TECPR2 | 14q32.31 | Autosomálně recesivní | Dětství | |
| SPG50 | 612936 | AP4M1 | 7q22.1 | Autosomálně recesivní | Dětství | |
| SPG51 | 613744 | AP4E1 | 15q21.2 | Autosomálně recesivní | Dětství | |
| SPG52 | 614067 | AP4S1 | 14q12 | Autosomálně recesivní | Dětství | |
| SPG53 | 614898 | VPS37A | 8p22 | Autosomálně recesivní | Dětství | |
| SPG54 | 615033 | DDHD2 | 8p11.23 | Autosomálně recesivní | Dětství | |
| SPG55 | 615035 | C12orf65 | 12q24.31 | Autosomálně recesivní | Dětství | |
| SPG56 | 615030 | CYP2U1 | 4q25 | Autosomálně recesivní | Dětství | |
| SPG57 | 615658 | TFG | 3q12.2 | Autosomálně recesivní | Brzy | |
| SPG58 | 611302 | KIF1C | 17p13.2 | Autosomálně recesivní | Během prvních dvou desetiletí | Spastická ataxie 2 |
| SPG59 | 603158 | USP8 | 15q21.2 | ? Autosomálně recesivní | Dětství | |
| SPG60 | 612167 | WDR48 | 3p22.2 | ? Autosomálně recesivní | Dětství | |
| SPG61 | 615685 | ARL6IP1 | 16p12.3 | Autosomálně recesivní | Dětství | |
| SPG62 | 615681 | ERLIN1 | 10q24.31 | Autosomálně recesivní | Dětství | |
| SPG63 | 615686 | AMPD2 | 1p13.3 | Autosomálně recesivní | Dětství | |
| SPG64 | 615683 | ENTPD1 | 10q24.1 | Autosomálně recesivní | Dětství | |
| SPG66 | 610009 | ARSI | 5q32 | Autosomálně dominantní | Dětství | |
| SPG67 | 615802 | PGAP1 | 2q33.1 | Autosomálně recesivní | Dětství | |
| SPG68 | 609541 | KLC2 | 11q13.1 | Autosomálně recesivní | Dětství | SPOAN syndrom |
| SPG69 | 609275 | RAB3GAP2 | 1q41 | Autosomálně recesivní | Dětství | Martsolfův syndrom, Warburgův mikro syndrom |
| SPG70 | 156560 | MARS | 12q13 | Autosomálně dominantní | Dětství | |
| SPG71 | 615635 | ZFR | 5p13.3 | ? Autosomálně recesivní | Dětství | |
| SPG72 | 615625 | REEP2 | 5q31 | Autozomálně recesivní; autosomálně dominantní | Dětství | |
| SPG73 | 616282 | CPT1C | 19q13.33 | Autozomálně dominantní | Dospělý | |
| SPG74 | 616451 | IBA57 | 1q42.13 | Autosomálně recesivní | Dětství | |
| SPG75 | 616680 | MAG | 19q13.12 | Autosomálně recesivní | Dětství | |
| SPG76 | 616907 | CAPN1 | 11q13 | Autosomálně recesivní | Dospělý | |
| SPG77 | 617046 | FARS2 | 6p25 | Autosomálně recesivní | Dětství | |
| SPG78 | 617225 | ATP13A2 | 1p36 | Autosomálně recesivní | Dospělý | Kufor – Rakebův syndrom |
| SPG79 | 615491 | UCHL1 | 4p13 | Autosomálně recesivní | Dětství | |
| HSNSP | 256840 | CCT5 | 5p15.2 | Autosomálně recesivní | Dětství | Dědičná senzorická neuropatie se spastickou paraplegií |
| SPG? | SERAC1 | 6q25.3 | Mladistvý | MEGDEL syndrom | ||
| SPG? | 605739 | KY | 3q22.2 | Autosomálně recesivní | Dětství | |
| SPG? | PLA2G6 | 22q13.1 | Autosomálně recesivní | Dětství | ||
| SPG? | ATAD3A | 1p36,33 | Autozomálně dominantní | Dětství | Harel-Yoonův syndrom | |
| SPG? | KCNA2 | 1p13.3 | Autozomálně dominantní | Dětství | ||
| SPG? | Granulin | 17q21.31 | ||||
| SPG? | POLR3A | 10q22.3 | Autosomálně recesivní |
Patofyziologie
Hlavním rysem HSP je axonální degenerace závislá na délce.[19] Patří mezi ně zkřížené a nezkřížené kortikospinální trakty na nohy a fasciculus gracilis. The spinocerebelární trakt je zapojen v menší míře. Neuronální buněčná těla degenerujících axonů jsou zachována a neexistují žádné důkazy o primárních demyelinizace.[16] V některých případech je pozorována ztráta buněk předního rohu míchy. Ganglie hřbetních kořenů, zadní kořeny a periferní nervy nejsou přímo ovlivněny.[Citace je zapotřebí ]
HSP ovlivňuje několik cest v motorických neuronech. Bylo identifikováno mnoho genů spojených s HSP. Výzvou zůstává přesně definovat klíčové hráče v každé z postižených drah, hlavně proto, že mnoho genů má více funkcí a je zapojeno do více než jedné cesty (obrázek).
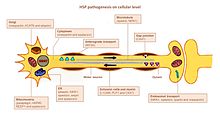
Hledání axonů
Hledání cesty je důležité pro růst axonů do správného cíle (např. Jiné nervové buňky nebo svalu). Pro tento mechanismus je významný gen L1CAM, buněčný povrchový glykoprotein nadrodiny imunoglobulinů. Mutace vedoucí ke ztrátě funkce v L1CAM se také vyskytují u jiných syndromů vázaných na X. Všechny tyto poruchy vykazují poškození kortikospinálního traktu (charakteristický znak HSP). L1CAM se účastní řady interakcí, váže se na další molekuly L1CAM a také na extracelulární buněčné adhezní molekuly, integriny a proteoglykany nebo intracelulární proteiny jako ankyriny.[Citace je zapotřebí ]
K defektu hledání cesty dochází prostřednictvím asociace L1CAM s neuropilin-1. Neuropilin-1 interaguje s proteiny Plexin-A za vzniku Semaforin-3A receptorový komplex. Semaphorin-a3A se poté uvolní do ventrální míchy, aby řídil kortikospinální neurony pryč od spojení míchy a míchy mezi středy. Pokud L1CAM nepracuje správně kvůli mutaci, neurony kortikospinálního systému nejsou nasměrovány do správné polohy a dojde k poškození.[3]
Metabolismus lipidů
Axony v centrálním a periferním nervovém systému jsou potaženy izolací, vrstvou myelinu, aby se zvýšila rychlost šíření akčního potenciálu. U některých forem hsp HSP je detekována abnormální myelinace v CNS.[20] Několik genů bylo spojeno s malformací myelinu, konkrétně PLP1, GFC2 a FA2H.[3] Mutace mění složení, tloušťku a integritu myelinu.[Citace je zapotřebí ]
Endoplazmatické retikulum (ER) je hlavní organelou pro syntézu lipidů. Mutace v genech kódujících proteiny, které mají roli při formování morfologie ER a metabolismu lipidů, byly spojeny s HSP. Mutace v ATL1, BSCL2 a ERLIN2 měnit strukturu ER, konkrétně trubicovou síť a tvorbu třícestných spojů v tubulech ER. Mnoho mutovaných genů souvisí s abnormálním metabolizmem lipidů. Nejčastější účinek je na kyselinu arachidonovou (CYP2U1 ) a cholesterol (CYP7B1 ) metabolismus, aktivita fosfolipázy (DDHD1 a DDHD2), tvorba gangliozidu (B4GALNT-1 ) a rovnováhu mezi metabolismem sacharidů a tuků (SLV33A1).[3][21][20]
Endosomální obchodování
Neurony přijímají látky ze svého okolí endocytózou. Endocytické vezikuly fúzují s endozomy za účelem uvolnění jejich obsahu. Existují tři hlavní oddíly, které mají obchodování s endosomy: Golgi do / z endosomů; plazmatická membrána do / z časných endosomů (prostřednictvím recyklace endosomů) a pozdních endosomů do lysozomů. Dysfunkce endosomálního obchodování může mít závažné následky u motorických neuronů s dlouhými axony, jak uvádí HSP. Mutace v AP4B1 a KIAA0415 jsou spojeny s narušením tvorby vezikul a transportem membrány, včetně selektivního vychytávání proteinů do vezikul. Oba geny kódují proteiny, které interagují s několika dalšími proteiny a narušují sekreční a endocytické cesty.[20]
Mitochondriální funkce
Mitochondriální dysfunkce byly spojeny s vývojovými a degenerativními neurologickými poruchami. Pouze několik genů HSP kóduje mitochondriální proteiny. Dva mitochondriální rezidentní proteiny jsou mutovány v HSP: paraplegin a chaperonin 60. Paraplegin je m-AAA metaloproteáza vnitřní mitochondriální membrány. Funguje při ribozomální montáži a kontrole kvality bílkovin. Zhoršená aktivita chaperoninu 60 vede ke zhoršené kontrole kvality mitochondrií. Dva geny DDHD1 a CYP2U1 ukázaly změnu mitochondriální architektury u pacientů s fibroblasty. Tyto geny kódují enzymy podílející se na metabolismu mastných kyselin.[Citace je zapotřebí ]
Diagnóza
Počáteční diagnóza HSP závisí na rodinné anamnéze, přítomnosti nebo nepřítomnosti dalších příznaků a vyloučení jiných negenetických příčin spasticity, což je zvláště důležité ve sporadických případech.[7]
Mozkové a míšní MRI je důležitý postup prováděný za účelem vyloučení dalších častých neurologických stavů, jako je roztroušená skleróza, ale také k detekci souvisejících abnormalit, jako je mozeček nebo corpus callosum atrofie stejně jako bílá hmota abnormality. Diferenciální diagnostika HSP by měl také vyloučit spastická diplegie který má téměř totožné každodenní účinky a je dokonce léčitelný podobnými léky, jako je baklofen a ortopedická operace; občas mohou tyto dvě podmínky vypadat a vypadat tak podobně, že jediné vnímáno rozdílem může být dědičná povaha HSP ve srovnání s výslovně nedědičnou povahou spastické diplegie (na rozdíl od spastické diplegie a jiných forem spastická mozková obrna, HSP nelze spolehlivě léčit selektivní dorzální rizotomie ).
Konečné potvrzení diagnózy HSP lze poskytnout pouze provedením genetické testy cílené na známé genetické mutace.
Klasifikace
Dědičné spastické paraplegie lze klasifikovat na základě příznaků; způsob dědičnosti; věk pacienta při nástupu; postižené geny; a zapojené biochemické cesty.
Léčba
Není známa žádná specifická léčba, která by zabránila, zpomalila nebo zvrátila HSP. Dostupné terapie spočívají hlavně v symptomatickém lékařském managementu a podpoře fyzické a emoční pohody. Terapeutika nabízená pacientům s HSP zahrnují:
- Baclofen - dobrovolný svalový relaxant k uvolnění svalů a snížení tónu. To může být podáváno orálně nebo intratekálně. (Studie na HSP [22][23][24])
- Tizanidin - k léčbě noční nebo přerušované křeče (studie k dispozici [25][26])
- Diazepam a klonazepam - snížit intenzitu křečí
- Oxybutyninchlorid - mimovolní svalové relaxanci a spazmolytikum, které se používá ke snížení spasticity močového měchýře u pacientů s problémy s ovládáním močového měchýře
- Tolterodin tartrát - mimovolní svalová relaxancia a spazmolytická látka, která se používá ke snížení spasticity močového měchýře u pacientů s problémy s ovládáním močového měchýře
- Systém Cro - ke snížení nadměrné svalové aktivity (stávající studie spasticity [27][28][29])
- Botulotoxin - ke snížení nadměrné svalové aktivity (stávající studie u pacientů s HSP[30][31])
- Antidepresiva (jako jsou selektivní inhibitory zpětného vychytávání serotoninu, tricyklická antidepresiva a inhibitory monoaminooxidázy) - pro pacienty s klinickou depresí
- Fyzikální terapie - obnovit a udržovat schopnost pohybu; snížit svalový tonus; k udržení nebo zlepšení rozsahu pohybu a mobility; zvýšit sílu a koordinaci; k prevenci komplikací, jako jsou zmrzlé klouby, kontraktury nebo proleženiny.
Prognóza
Ačkoli HSP je progresivní stav, prognóza u jedinců s HSP se velmi liší. Primárně postihuje nohy, i když u některých jedinců může docházet k určitému zapojení horní části těla. Některé případy se vážně deaktivují, zatímco jiné méně deaktivují a jsou kompatibilní s produktivním a plnohodnotným životem. Většina jedinců s HSP má normální stav délka života.[14]
Epidemiologie
Po celém světě se prevalence všech dědičných spastických paraplegií dohromady odhaduje na 2 až 6 ze 100 000 lidí.[32] A Norština Studie s více než 2,5 miliony lidí publikovaná v březnu 2009 zjistila míru prevalence HSP 7,4 / 100 000 populace - vyšší míru, ale ve stejném rozsahu jako předchozí studie. Nebyly nalezeny žádné rozdíly v míře týkající se pohlaví a průměrný věk při nástupu byl 24 let.[33] V Spojené státy „Dědičná spastická paraplegie je uvedena jako„ vzácná nemoc “Úřadem pro vzácné nemoci (ORD) Národní institut zdraví což znamená, že porucha postihuje méně než 200 000 lidí v americké populaci.[32]
Reference
- ^ Fink, John K. (01.08.2003). „Dědičné spastické paraplegie: devět genů a počítání“. Archivy neurologie. 60 (8): 1045–1049. doi:10.1001 / archneur.60.8.1045. ISSN 0003-9942. PMID 12925358.
- ^ Depienne, Christel; Stevanin, Giovanni; Brice, Alexis; Durr, Alexandra (01.12.2007). "Dědičné spastické paraplegie: aktualizace". Aktuální názor na neurologii. 20 (6): 674–680. doi:10.1097 / WCO.0b013e3282f190ba. ISSN 1350-7540. PMID 17992088. S2CID 35343501.
- ^ A b C d Blackstone, Craig (21. července 2012). "Buněčné cesty dědičné spastické paraplegie". Roční přehled neurovědy. 35 (1): 25–47. doi:10.1146 / annurev-neuro-062111-150400. PMC 5584684. PMID 22540978.
- ^ De Matteis, Maria Antonietta; Luini, Alberto (08.09.2011). „Mendelovy poruchy obchodování s membránami“. The New England Journal of Medicine. 365 (10): 927–938. doi:10.1056 / NEJMra0910494. ISSN 1533-4406. PMID 21899453. S2CID 14772080.
- ^ Faber I, Pereira ER, Martinez AR, França M Jr, Teive HA (listopad 2013). „Dědičná spastická paraplegie od roku 1880 do roku 2017: historický přehled“. Arquivos de Neuro-Psiquiatria. Brazilská neurologická akademie. 75 (11): 813–818. doi:10.1590 / 0004-282X20170160. PMID 29236826.
- ^ Lorrain, Maurice. Příspěvek à l'étude de la paraplégie spasmodique familiale: travail de la clinique des maladies du système nervux à la Salpêtrière. G. Steinheil, 1898.
- ^ A b C d Harding, AE (1983). "Klasifikace dědičných ataxií a paraplegií". Lanceta. New York: Lancet. 1 (8334): 1151–5. doi:10.1016 / s0140-6736 (83) 92879-9. PMID 6133167. S2CID 6780732.
- ^ McAndrew CR, Harms P (2003). „Parestezie při kombinované spinální epidurální injekci jehlou přes jehlu versus jednorázová páteř pro elektivní císařský řez“. Anestezie a intenzivní péče. 31 (5): 514–517. doi:10.1177 / 0310057X0303100504. PMID 14601273.
- ^ Fjermestad, Krister W .; Kanavin, Øivind J .; Næss, Eva E .; Hoxmark, Lise B .; Hummelvoll, Grete (13. 7. 2016). „Zdravotní průzkum dospělých s dědičnou spastickou paraparézou ve srovnání s kontrolami populační studie“. Orphanet Journal of Rare Diseases. 11 (1): 98. doi:10.1186 / s13023-016-0469-0. ISSN 1750-1172. PMC 4944497. PMID 27412159.
- ^ Chaudhuri, Abhijit; Behan, Peter O. (2004-03-20). „Únava při neurologických poruchách“. Lanceta. 363 (9413): 978–988. doi:10.1016 / S0140-6736 (04) 15794-2. ISSN 1474-547X. PMID 15043967. S2CID 40500803.
- ^ „Dědičná spastická paraplegie“. nhs.uk. 2017-10-18. Citováno 2018-01-28.
- ^ Fink JK (2003). „Dědičná spastická paraplegie“. Archivy neurologie. 60 (8): 1045–1049. doi:10.1001 / archneur.60.8.1045. PMID 12925358.
- ^ A b C Harding AE (1981). „Dědičná“ čistá „spastická paraplegie: klinická a genetická studie 22 rodin“. Časopis neurologie, neurochirurgie a psychiatrie. 44 (10): 871–883. doi:10.1136 / jnnp.44.10.871. PMC 491171. PMID 7310405.
- ^ A b Depienne C, Stevanin G, Brice A, Durr A (2007). „Hereditary Spastic Paraplegia: An Update“. Aktuální názor na neurologii. 20 (6): 674–680. doi:10.1097 / WCO.0b013e3282f190ba. PMID 17992088. S2CID 35343501.
- ^ A b C Schüle, Rebecca; Wiethoff, Sarah; Martus, Peter; Karle, Kathrin N .; Otto, Susanne; Klebe, Stephan; Klimpe, Sven; Gallenmüller, Constanze; Kurzwelly, Delia (01.04.2016). „Dědičná spastická paraplegie: klinicko-genetické lekce od 608 pacientů“. Annals of Neurology. 79 (4): 646–658. doi:10.1002 / ana.24611. ISSN 1531-8249. PMID 26856398. S2CID 10558032.
- ^ A b Schüle R, Schöls L (2011) Genetika dědičných spastických paraplegií. Semin Neurol 31 (5): 484-493
- ^ Wang YG, Shen L (2009) AAA ATPázy a dědičná spastická paraplegie. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 26 (3): 298-301
- ^ Helbig KL, Hedrich UB, Shinde DN, Krey I, Teichmann AC, Hentschel J, Schubert J, Chamberlin AC, Huether R, Lu HM4, Alcaraz WA, Tang S, Jungbluth C, Dugan SL, Vainionpää L, Karle KN, Synofzik M , Schöls L, Schüle R, Lehesjoki AE, Helbig I, Lerche H, Lemke JR (2016) Recidivující mutace v KCNA2 jako nová příčina dědičné spastické paraplegie a ataxie. Ann Neurol 80 (4)
- ^ Wharton SB, McDermott CJ, Grierson AJ, Wood JD, Gelsthorpe C, Ince PG, Shaw PJ (2003) Buněčná a molekulární patologie motorického systému při dědičné spastické paraparéze v důsledku mutace genu spastinu. J Neuropathol Exp Neurol 62: 1166–1177
- ^ A b C Noreau, A., Dion, P.A. & Rouleau, G.A., 2014. Molekulární aspekty dědičné spastické paraplegie. Experimental Cell Research, 325 (1), s. 18–26
- ^ Lo Giudice, T. a kol., 2014. Dědičná spastická paraplegie: Klinicko-genetické vlastnosti a vyvíjející se molekulární mechanismy. Experimental Neurology, 261, str. 518–539.
- ^ Margetis K, Korfias S, Boutos N, Gatzonis S, Themistocleous M, Siatouni A a kol. Intratekální léčba baklofenem pro symptomatickou léčbu dědičné spastické paraplegie. Klinická neurologie a neurochirurgie. 2014; 123: 142-5.
- ^ Heetla HW, Halbertsma JP, Dekker R, Staal MJ, van Laar T. Zlepšil výkon v chůzi u pacienta s dědičnou spastickou paraplegií po kontinuální intratekální infuzi baklofenového testu a následné implantaci pumpy: kazuistika. Archivy fyzikální medicíny a rehabilitace. 2015; 96 (6): 1166-9.
- ^ Klebe S, Stolze H, Kopper F, Lorenz D, Wenzelburger R, Deuschl G a kol. Objektivní hodnocení chůze po intratekálním baklofenu v dědičné spastické paraplegii. Journal of Neurology. 2005; 252 (8): 991-3.
- ^ Knutsson E, Mårtensson A, Gransberg L. Antiparetické a antispastické účinky indukované tizanidinem u pacientů se spastickou parézou. Časopis neurologických věd. 1982; 53 (2): 187-204.
- ^ Bes A, Eyssette M, Pierrot-Deseilligny E, Rohmer F, Warter JM. Multicentrická, dvojitě zaslepená studie s tizanidinem, novým antispastickým činidlem, se spasticitou spojenou s hemiplegií. Současný lékařský výzkum a názory. 1988; 10 (10): 709-18.
- ^ Celletti C, Camerota F. Předběžný důkaz účinků vibrací fokálních svalů na spasticitu způsobenou mozkovou obrnou u malého vzorku italských dětí. Clin Ter. 162 (5): 125-8. 2011
- ^ Caliandro P, Celletti C, Padua L, Minciotti I, Russo G, Granata G, La Torre G, Granieri E, Camerota F. Vibrace fokálního svalu při léčbě spasticity horních končetin: pilotní randomizovaná kontrolovaná studie u pacientů s chronickou cévní mozkovou příhodou. Arch Phys Med Rehabil. 93 (9): 1656-1661. 2012.
- ^ . Casale R1, Damiani C, Maestri R, Fundarò C, Chimento P, Foti C. Zaostřená vibrace 100 Hz zlepšuje funkci a snižuje spasticitu horní končetiny: dvojitě zaslepená kontrolovaná studie. Eur J Phys Rehabil Med. Říjen 2014; 50 (5): 495-504. 2014.
- ^ Hecht MJ, Stolze H, Auf Dem Brinke M, Giess R, Treig T, Winterholler M a kol. Injekce botulotoxinu typu A snižují spasticitu u mírné až středně závažné dědičné spastické paraplegie - zpráva o 19 případech. Poruchy pohybu. 2008; 23 (2): 228-33.
- ^ de Niet M, de Bot ST, van de Warrenburg BP, Weerdesteyn V, Geurts AC. Funkční účinky léčby botulotoxinem typu A a následné protažení spastických lýtkových svalů: Studie u pacientů s dědičnou spastickou paraplegií. Journal of rehabilitation medicine. 2015; 47 (2): 147-53.
- ^ A b Národní institut zdraví (2008). „Hereditary Spastic Paraplegia Information Page“. Archivovány od originál dne 2014-02-21. Citováno 2008-04-30.
- ^ Erichsen, AK; Koht, J; Stray-Pedersen, A; Abdelnoor, M; Tallaksen, CM (červen 2009). „Prevalence dědičné ataxie a spastické paraplegie v jihovýchodním Norsku: populační studie“ (PDF). Mozek. 132 (Pt 6): 1577–1888. doi:10.1093 / brain / awp056. PMID 19339254.
Další čtení
- GeneReviews / NCBI / NIH / UW vstup na Spastic Paraplegia 3A
- GeneReviews / NCBI / NIH / UW záznam v přehledu Hereditary Spastic Paraplegia
- Warner, Tom (leden – únor 2007). „Dědičná spastická paraplegie“ (PDF). Pokroky v klinické neurovědě a rehabilitaci. 6 (6): 16–17.
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |