DiGeorgeův syndrom - DiGeorge syndrome
| DiGeorgeův syndrom | |
|---|---|
| Ostatní jména | DiGeorge anomálie,[1][2] velokardiofaciální syndrom (VCFS),[3] Shprintzenův syndrom,[4] syndrom obličejové anomálie (CTAF),[5] Takao syndrom,[6] Sedlackova syndrom,[7] Caylerův kardiofaciální syndrom,[7] CATCH22,[7] Syndrom delece 22q11.2[7] |
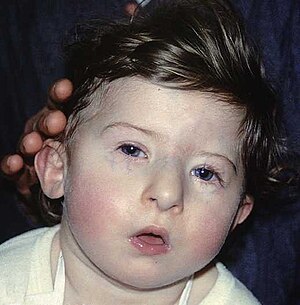 | |
| Dítě s charakteristickými rysy obličeje DiGeorgeova syndromu | |
| Specialita | Lékařská genetika |
| Příznaky | Pestrý; běžně vrozené srdeční problémy, specifické rysy obličeje, rozštěp patra[7] |
| Komplikace | Problémy s ledvinami, ztráta sluchu, autoimunitní poruchy[7] |
| Příčiny | Genetický (typicky nová mutace)[7] |
| Diagnostická metoda | Na základě příznaků a genetické testování[5] |
| Diferenciální diagnostika | Smith – Lemli – Opitzův syndrom, Alagilleův syndrom, VACTERL, Okulo-auriculo-vertebrální spektrum[5] |
| Léčba | Zahrnuje mnoho specializací v oblasti zdravotní péče[5] |
| Prognóza | Závisí na konkrétních příznacích[3] |
| Frekvence | 1 ze 4 000[7] |
DiGeorgeův syndrom, také známý jako Syndrom delece 22q11.2, je syndrom způsobený delecí malého segmentu chromozom 22.[7] I když se příznaky mohou lišit, často zahrnují vrozené srdeční problémy, specifické rysy obličeje, časté infekce, vývojové zpoždění, problémy s učením a rozštěp patra.[7] Přidružené podmínky zahrnují problémy s ledvinami, ztráta sluchu a autoimunitní poruchy jako revmatoidní artritida nebo Gravesova nemoc.[7]
DiGeorgeův syndrom je obvykle způsoben vypuštěním 30 až 40 geny uprostřed chromozom 22 v a umístění známý jako 22q11.2.[3] Asi 90% případů nastává kvůli novému mutace během raného vývoje, zatímco 10% je zdědil od rodičů člověka.[7] to je autosomálně dominantní, což znamená, že ke vzniku onemocnění je zapotřebí pouze jeden ovlivněný chromozom.[7] Diagnóza je podezřelá na základě příznaků a potvrzena genetické testování.[5]
Ačkoli neexistuje léčba, léčba může zlepšit příznaky.[3] To často zahrnuje a multidisciplinární přístup se snahou o zlepšení funkce potenciálně mnoha zapojených orgánových systémů.[8] Dlouhodobé výsledky závisí na přítomných příznacích a závažnosti problémů se srdcem a imunitním systémem.[3] Při léčbě může být průměrná délka života normální.[9]
DiGeorgeův syndrom se vyskytuje asi u 1 ze 4 000 lidí.[7] Syndrom byl poprvé popsán v roce 1968 americkým lékařem Angelo DiGeorge.[10][11] Na konci roku 1981 byla stanovena základní genetika.[11]
Příznaky a symptomy
Rysy tohoto syndromu se velmi liší, dokonce iu členů stejné rodiny, a ovlivňují mnoho částí těla. Charakteristické příznaky mohou zahrnovat vrozené vady, jako jsou vrozená srdeční onemocnění, vady patra, nejčastěji spojené s neuromuskulárními problémy s uzávěrem (velofaryngeální nedostatečnost ), poruchy učení, mírné rozdíly v obličejových vlastnostech a opakující se infekce. Infekce jsou u dětí časté kvůli problémům s imunitní systém je T buňka -zprostředkovaná odpověď že u některých pacientů je v důsledku chybějící nebo hypoplastický brzlík. DiGeorgeův syndrom může být poprvé spatřen, když má postižený novorozenec srdeční vady nebo křeče hypokalcémie kvůli poruše příštítných tělísek a nízké hladiny parathormonu (parathormon ).
Postižení jedinci mohou mít jako kojenci také jiné druhy vrozených vad, včetně abnormalit ledvin a významných potíží s krmením. U této populace pacientů jsou také velmi časté gastrointestinální problémy. Problémy s trávicí pohyblivostí mohou vést k zácpě.[12] Poruchy jako např hypotyreóza a hypoparatyreóza nebo trombocytopenie (nízké hladiny krevních destiček) a psychiatrické nemoci jsou běžné pozdně se vyskytující rysy.[13]
Mikrodelece v chromozomální oblasti 22q11.2 jsou spojeny s 20 až 30krát vyšším rizikem schizofrenie.[14] Studie poskytují různé míry 22q11,2DS u schizofrenie, v rozmezí od 0,5 do 2,0% a v průměru kolem 1,0%, ve srovnání s celkovým odhadovaným 0,025% rizikem 22q11.2DS v obecné populaci.[15]
Hlavní rysy lze shrnout pomocí mnemotechnické pomůcky ÚLOVEK-22 popsat 22q11.2DS, přičemž 22 značící chromozomální abnormalitu lze nalézt na 22. chromozomu, jak je uvedeno níže:[16]
- Srdeční abnormality (obvykle přerušený aortální oblouk, truncus arteriosus a Fallotova tetralogie )
- Abnormální facie
- Thymic aplázie
- Rozštěp patra
- Hypokalcemie / hypoparatyreóza
Jednotlivci mohou mít mnoho možných funkcí, od počtu souvisejících funkcí, od mírných po velmi závažné. Mezi běžné příznaky patří:
- Vrozená srdeční choroba (40% jednotlivců), zejména conotruncal malformace (přerušený aortální oblouk (50%), perzistentní truncus arteriosus (34%), Fallotova tetralogie a defekt komorového septa )
- Cyanóza (namodralá kůže kvůli špatnému oběhu krve bohaté na kyslík)
- Palatal abnormality (50%), zvláště velofaryngeální neschopnost submukózně rozštěp patra, a rozštěp patra; charakteristické rysy obličeje (přítomné ve většině kavkazský jednotlivci) včetně hypertelorismus
- Problémy s učením (90%), včetně kognitivní deficity, poruchy pozornosti[17]
- Hypokalcémie (50%) (kvůli hypoparatyreóze)
- Významný krmení problémy (30%)
- Renální anomálie (37%)
- Ztráta sluchu (oba vodivý a senzorineurální ) (ztráta sluchu s kraniofaciálními syndromy )
- Laryngotracheoezofageální anomálie
- Růstový hormon nedostatek
- Autoimunitní poruchy
- Poruchy imunity kvůli snížené T buňka čísla
- Záchvaty (s nebo bez hypokalcémie )
- Kosterní abnormality
- Psychiatrické poruchy[17]
Tento syndrom je charakterizován neúplná penetrace. Proto existuje výrazná variabilita v klinické expresi mezi různými pacienty. To často ztěžuje včasnou diagnostiku.[18]
Kognitivní poruchy
Děti s DiGeorgeovým syndromem mají v neuropsychologických testech specifický profil. Obvykle mají podlimitní normální IQ, přičemž většina jednotlivců má vyšší skóre ve verbálních než neverbálních doménách. Někteří mohou navštěvovat běžné školy, zatímco jiní se vzdělávají doma nebo ve zvláštních třídách. Závažnost hypokalcemie v raném dětství je spojena s problémy s chováním podobným autismu.[19]
Dospělí s DiGeorgeovým syndromem jsou zvláště vysoce rizikovou skupinou pro rozvoj schizofrenie. Asi 30% má alespoň jeden incident z psychóza a zhruba čtvrtina se vyvíjí skutečně schizofrenie.[20]
Jedinci s DiGeorgeovým syndromem mají také vyšší riziko vzniku časného nástupu Parkinsonova choroba (PD). Diagnostiku Parkinsonovy choroby lze odložit až o 10 let antipsychotika, což může způsobit parkinsonské příznaky.[21][22]
Řeč a jazyk
Současný výzkum ukazuje, že s profilem 22q11.2DS je spojen jedinečný profil poruch řeči a jazyka. Děti často dosahují nižšího hodnocení řeči a jazyka ve srovnání s neverbálním skóre IQ.[rozporuplný ] Mezi běžné problémy patří nadnárodnost, jazykové zpoždění a zvukové chyby řeči.[23][24][25]
Nadnárodnost nastává, když vzduch uniká nosem během produkce zvuků ústní řeči, což vede ke snížení srozumitelnost. To je společná charakteristika řečového a jazykového profilu, protože to má 69% dětí palatal abnormality. Pokud je struktura měkkého patra velum je taková, že nezabrání proudění vzduchu nahoru do nosní dutina, způsobí to hypernazální řeč. Tento jev se označuje jako velofaryngeální nedostatečnost (VPI). Ztráta sluchu může také přispět ke zvýšené hypernazalitě, protože děti se sluchovým postižením mohou mít potíže s vlastním sledováním svého ústního projevu. Možnosti léčby dostupné pro VPI zahrnují protézu a chirurgický zákrok.[23][24][26][27][28]
Obtíže se získáním slovní zásoby a formulací mluveného jazyka (expresivní jazyk deficity) na počátku vývoje jazyka jsou také součástí řečového a jazykového profilu spojeného s odstraněním 22q11.2. Získávání slovní zásoby je u dětí předškolního věku často výrazně zpožděna. V některých nedávných studiích měly děti výrazně omezenou slovní zásobu nebo stále nebyly verbální ve věku 2–3 let. Děti ve školním věku dělají pokroky s expresivním jazykem, jak dospívají, ale mnoho z nich má i nadále zpoždění a projevuje potíže, když jim jsou předloženy jazykové úkoly, jako je verbální vybavování příběhů a vytváření delších a složitějších vět. Receptivní jazyk, což je schopnost porozumět, udržet si nebo zpracovat mluvený jazyk, může být také narušen, i když ne obvykle se stejnou závažností jako výrazové narušení jazyka.[24][27][28][29]
Artikulace chyby se běžně vyskytují u dětí s DiGeorgeovým syndromem. Mezi tyto chyby patří omezený fonemický (zvuk řeči) inventář a použití kompenzačních artikulačních strategií vedoucích ke snížené srozumitelnosti. The phonemic inventář, který se obvykle vyrábí, se skládá ze zvuků vydávaných v přední nebo zadní části ústní dutiny, například: / p /, / w /, / m /, / n / a rázy. Zvuk vydávaný uprostřed úst zcela chybí. Mezi kompenzační chyby artikulace této populace dětí patří: rázy se zastaví, nosní substituce, faryngální frikativy, lingvalalatální sykavky, snížený tlak na zvuky souhlásek nebo kombinace těchto příznaků. Z těchto chyb mají nejvyšší frekvenci rázů. Je odůvodněné, že omezené fonemický soupis a je zde použití kompenzačních artikulačních strategií kvůli strukturálním abnormalitám patra. Poruchy řeči, které tato populace vykazuje, jsou v mladším věku závažnější a vykazují trend postupného zlepšování, jak dítě dospívá.[23][27]
Genetika

DiGeorgeův syndrom je způsoben a heterozygotní delece části dlouhého ramene (q) chromozomu 22, oblast 1, pásmo 1, dílčí pásmo 2 (22q11.2). Přibližně 80-90% pacientů má deleci 3 Mb a 8% má vymazání 1,5 MB.[30][31] Počet genů ovlivněných delecí byl citován jako přibližně 30 až 50.[32][33] Velmi zřídka mohou mít pacienti s poněkud podobnými klinickými rysy delece na krátkém rameni chromozomu 10.[34] Porucha má autosomálně dominantní vzor dědičnosti.
Francouzská studie se 749 osobami diagnostikovanými v letech 1995 až 2013 zjistila, že mutace byla zděděna u 15% pacientů, z nichž 85,5% pocházelo od matky.[35] Jiné studie zjistily míru dědičnosti 6–10%. Většina případů je výsledkem a de novo (nové v rodině) vymazání.[12] Je to proto, že oblast 22q11 má strukturu, díky níž je vysoce náchylná k přeskupení během tvorby spermií nebo vajíček.[36]
Přesný mechanismus, který způsobuje všechny související rysy syndromu, není znám.[30] Z 30–50 genů v odstraněné oblasti bylo identifikováno mnoho z nich, které pravděpodobně hrají roli ve vývoji některých příznaků a příznaků.
TBX1
Štěstí nedostatek z TBX1 gen (transkripční faktor T-boxu TBX1) je považován za příčinu některých pozorovaných symptomů. Bodové mutace v tomto genu byly také pozorovány u jedinců s DiGeorgeovým syndromem.[30] TBX1 je součástí T-box rodina genů, které mají důležitou roli při tvorbě tkání a orgánů během embryonálního vývoje a mohou hrát roli v regulaci diferenciace po migraci buňky neurální lišty. Nervový hřeben tvoří mnoho struktur ovlivněných DiGeorgeovým syndromem, včetně kostí lebky, mezenchymu obličeje a patra, výtokového traktu srdce a brzlíku a příštítných tělísek stroma. Když dojde ke ztrátě výrazu FGF18 během vývoje hltanové oblouky je vidět smrt buněk nervového hřebenu. Ačkoli ani FGF18, ani TBX1 nejsou exprimovány v buňkách neurální lišty, TBX1 může mít roli v regulaci exprese FGF18, což zajišťuje správnost diferenciace těchto buněk v oblasti hltanu. Proto může být dysfunkce TBX1 zodpovědná za některé příznaky DiGeorgeova syndromu.[31]
Výzkum na myších modelech ukázal, že delece Tbx1 vede k několika defektům podobným těm, které se vyskytují u lidí, zejména ovlivňující vývoj velké tepny a brzlík.[37][38]
Abnormality pozorované ve velkých tepnách myší s deficitem Tbx1 jsou důsledkem abnormální tvorby a remodelace aortální oblouky během raného vývoje. Role Tbx1 pro správnou tvorbu a remodelaci aortálních oblouků byla rozsáhle studována na různých myších modelech, což naznačuje klíčovou roli Tbx1 pro kardiovaskulární vývoj a fenotypy pozorované u DiGeorgeova syndromu.
DGCR8
U myší je haploinsufficient DGCR8 Gen byl spojen s nesprávnou regulací mikroRNA miR-338 a 22q11.2 deleční fenotypy.[39]
TANGO2
Transport and golgi organization 2 homolog (TANGO2 ) také známý jako otevřený čtecí rámec 25 chromozomu 22 (C22orf25) je protein, který je u lidí kódován genem TANGO2.
Gen kódující C22orf25 je lokalizován na chromozomu 22 a místě q11.21, takže je často spojován s delečním syndromem 22q11.2.[40] Ale s poruchou TANGO2, která je autosomálně recesivní, nenastane ve všech případech.
Mutace v genu TANGO2 mohou způsobit defekty v mitochondriích β-oxidace[41] a zvýšil endoplazmatické retikulum stres a snížení Golgi objemová hustota.[42] Výsledkem těchto mutací je časný nástup hypoglykémie, hyperamonémie, rhabdomyolýza, srdeční arytmie, a encefalopatie které se později vyvinou v kognitivní poruchy.[41][42]
Geny pro Parkinsonovu chorobu
22q11.2DS byl spojován s vyšším rizikem časného nástupu Parkinsonova choroba (PD). Viděná neuropatologie je podobná LRRK2 spojené s PD. Žádný z genů ovlivněných u jedinců s 22q11.2DS nebyl dříve spojen s PD, ale existuje řada pravděpodobných kandidátů. Patří mezi ně DGCR8, který je důležitý pro biogenezi mozkové mikroDNA, SRPT5 který kóduje protein, který interaguje s PARK2 protein, COMT který se podílí na regulaci hladin dopaminu, a mikroRNA miR-185, o které se předpokládá, že cílí na známé PD lokusy LRRK2.[21]
Diagnóza


Diagnóza DiGeorgeova syndromu může být obtížná kvůli množství potenciálních příznaků a rozdílům ve fenotypech mezi jednotlivci. Je podezření na to u pacientů s jedním nebo více příznaky delece. V těchto případech je diagnóza 22q11.2DS potvrzena pozorováním delece části dlouhého ramene (q) chromozomu 22, oblast 1, pásmo 1, dílčí pásmo 2. Genetická analýza se obvykle provádí pomocí fluorescence in situ hybridizace (FISH), který je schopen detekovat mikrodelece, které standardní karyotypují (např. G-bandáž ) slečna, minout. Mezi novější metody analýzy patří Multiplexní ligace závislé zesílení sondy test (MLPA) a kvantitativní polymerázová řetězová reakce (qPCR), oba mohou detekovat atypické delece v 22q11.2, které nejsou detekovány FISH.[44] Analýza qPCR je také rychlejší než analýza FISH, která může mít obrat 3 až 14 dní.[12]
Studie z roku 2008 týkající se nové sondy MLPA s vysokým rozlišením vyvinuté k detekci variace počtu kopií při 37 bodech na chromozomu 22q zjistil, že je stejně spolehlivý jako FISH při detekci normálních delecí 22q11.2. Rovněž dokázala detekovat menší atypické delece, které se pomocí FISH snadno minou. Tyto faktory spolu s nižšími náklady a snazším testováním znamenají, že tato sonda MLPA by mohla nahradit FISH v klinickém testování.[45]
Genetické testování pomocí BACs-on-Beads bylo úspěšné při detekci delecí v souladu s 22q11.2DS během prenatálního testování.[46][47] Genomická hybridizace srovnávající pole (array-CGH) používá velké množství sond embosovaných v čipu k screeningu celého genomu na delece nebo duplikace. Může být použit v postnatální a prenatální diagnostice 22q11.2.[48]
Méně než 5% jedinců s příznaky DiGeorgeova syndromu má normální rutinní cytogenetické studie a negativní testování FISH. V těchto případech jsou příčinou atypické delece.[49] Některé případy syndromu delece 22q11.2 mají defekty v jiných chromozomech, zejména deleci v chromozomové oblasti 10p14.[34]
Léčba
Pro DiGeorgeův syndrom není známa žádná léčba. Některé jednotlivé funkce lze léčit pomocí standardního ošetření.[50] Klíčem je identifikovat každou z přidružených funkcí a spravovat každou pomocí nejlepší dostupné léčby.
Například u dětí je důležité včas identifikovat problémy s imunitou, protože jsou nutná zvláštní opatření týkající se transfuze krve a imunizace živými vakcínami.[51] Transplantace brzlíku lze použít k řešení nepřítomnosti brzlíku u vzácného, takzvaného „úplného“ DiGeorgeova syndromu.[52] Bakteriální infekce jsou léčeni antibiotika. Operace srdce je často vyžadována pro vrozené srdeční vady. Hypoparatyreóza způsobující hypokalcemii často vyžaduje celoživotní doplňky vitaminu D a vápníku. Specializované kliniky, které poskytují vícesystémovou péči, umožňují jednotlivcům s DiGeorgeovým syndromem vyhodnotit všechny jejich zdravotní potřeby a umožňují pečlivé sledování pacientů. Příkladem tohoto typu systému je 22q Deletion Clinic at Nemocnice SickKids v kanadském Torontu, který poskytuje dětem průběžnou podporu syndromu delece 22q11, lékařskou péči a informace od týmu zdravotnických pracovníků.[53]
Epidemiologie
Odhaduje se, že DiGeorgeův syndrom postihuje mezi jedním v roce 2000 a jedním ze 4000 živě narozených dětí.[54][55] Tento odhad je založen na hlavních vrozených vadách a může být podceňován, protože někteří jedinci s delecí mají málo příznaků a nemusí být formálně diagnostikováni. Je to jedna z nejčastějších příčin mentální postižení kvůli syndromu genetické delece.[56]
Očekává se, že počet postižených osob vzroste z několika důvodů: (1) chirurgický a lékařský pokrok, stále více lidí přežívá srdeční vady spojené se syndromem. Tito jedinci zase mají děti. Šance, že osoba s DiGeorgeovým syndromem bude mít postižené dítě, je 50% za každé těhotenství; (2) Rodiče, kteří zasáhli děti, ale nevěděli o svých genetických podmínkách, jsou nyní diagnostikováni, jakmile budou k dispozici genetické testy; (3) Techniky molekulární genetiky, jako je FISH (fluorescence in situ hybridizace), mají svá omezení a nebyly schopny detekovat všechny delece 22q11.2. Novější technologie dokázaly tyto atypické delece detekovat.[57]
název
Známky a příznaky DiGeorgeova syndromu jsou tak rozmanité, že různá seskupení jeho vlastností byla kdysi považována za samostatné podmínky. Tyto původní klasifikace zahrnovaly velokardiofaciální syndrom, Shprintzenův syndrom, DiGeorgeovu sekvenci / syndrom, Sedlackovův syndrom a syndrom obličeje anomálie tváře. Všechny jsou nyní chápány jako projevy jediného syndromu.
Verze ICD-10 2015 zmiňuje DiGeorgeův syndrom pomocí dvou kódů: D82.1 (Di Georgeův syndrom)[58] a Q93.81 (Velo-kardio-obličejový syndrom).[59] Návrh ICD-11 Beta pojednává o syndromu pod „fenotypem LD50.P1 CATCH 22“.[59] Protože však tento syndrom je způsoben vypuštěním malého kousku chromozom 22, někteří doporučují, aby byl použit název „22q11.2 deleční syndrom (22q11.2DS)“.[60][12] Někteří odborníci podporují změnu názvu jak DiGeorge, tak velocardiofacial syndromy na CATCH-22.[Citace je zapotřebí ] Mezinárodní nadace 22q11.2 se prostřednictvím své „kampaně se stejným názvem“ zasazuje o syndrom vymazání názvu 22q11.2.[61]
Viz také
Reference
- ^ Rapini, Ronald P .; Bolognia, Jean L .; Jorizzo, Joseph L. (2007). Dermatologie: dvoudílná sada. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- ^ James, William D .; Berger, Timothy G .; et al. (2006). Andrewsovy nemoci kůže: klinická dermatologie. Saunders Elsevier. ISBN 978-0-7216-2921-6.
- ^ A b C d E „Syndrom delece 22q11.2“. Informační centrum o genetických a vzácných onemocněních (GARD). Archivováno z původního dne 5. července 2017. Citováno 15. května 2017.
- ^ Shprintzen RJ, Goldberg RB, Lewin ML, Sidoti EJ, Berkman MD, Argamaso RV, Young D (leden 1978). „Nový syndrom zahrnující rozštěp patra, srdeční anomálie, typické facie a poruchy učení: velo-kardio-obličejový syndrom“. Rozštěp patra J.. 15 (1): 56–62. PMID 272242.
- ^ A b C d E „Syndrom delece chromozomu 22q11.2 - NORD (Národní organizace pro vzácné poruchy)“. NORD (Národní organizace pro vzácné poruchy). 2017. Archivováno z původního dne 28. ledna 2017. Citováno 10. července 2017.
- ^ Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, Scambler P, Goodship J (říjen 1993). „Syndrom tváře anotie anotie je spojen s delecí v chromozomu 22q11“. J. Med. Genet. 30 (10): 822–4. doi:10,1136 / jmg.30.10.822. PMC 1016562. PMID 8230157.
- ^ A b C d E F G h i j k l m n „Syndrom delece 22q11.2“. Genetická domácí reference. Červenec 2013. Archivováno z původního dne 13. května 2017. Citováno 15. května 2017.
- ^ Kobrynski LJ, Sullivan KE (říjen 2007). "Velocardiofacial syndrom, DiGeorgeův syndrom: syndromy delece chromozomu 22q11.2". Lanceta. 370 (9596): 1443–52. doi:10.1016 / S0140-6736 (07) 61601-8. PMID 17950858.
- ^ Goldman, Lee; Schafer, Andrew I. (2015). E-kniha o medicíně Goldman-Cecil. Elsevier Health Sciences. str. 702. ISBN 9780323322850. Archivováno od originálu dne 2017-11-05.
- ^ DiGeorge, A (1968). „Vrozená absence brzlíku a jeho imunologické důsledky: souběh s vrozenou hypoparatyreoidismem“. March of Dimes-Defects Defects Foundation: 116–21.
- ^ A b Restivo A, Sarkozy A, Digilio MC, Dallapiccola B, Marino B (únor 2006). „Deleční syndrom 22q11: přehled některých aspektů vývojové biologie kardiovaskulárního systému“. J Cardiovasc Med (Hagerstown). 7 (2): 77–85. doi:10.2459 / 01.JCM.0000203848.90267.3e. PMID 16645366.
- ^ A b C d McDonald-McGinn DM, Sullivan KE (leden 2011). „Syndrom delece chromozomu 22q11.2 (DiGeorgeův syndrom / velocardiofacial syndrom)“. Medicína (Baltimore). 90 (1): 1–18. doi:10.1097 / MD.0b013e3182060469. PMID 21200182.
- ^ Debbané M, Glaser B, David MK, Feinstein C, Eliez S (2006). „Psychotické příznaky u dětí a dospívajících se syndromem delece 22q11.2: neuropsychologické a behaviorální důsledky“. Schizophr. Res. 84 (2–3): 187–93. doi:10.1016 / j.schres.2006.01.019. PMID 16545541.
- ^ [není nutný primární zdroj ] Bassett AS, Chow EW, AbdelMalik P, Gheorghiu M, Husted J, Weksberg R (2003). „Fenotyp schizofrenie u syndromu delece 22q11“. Jsem J. Psychiatrie. 160 (9): 1580–6. doi:10.1176 / appi.ajp.160.9.1580. PMC 3276594. PMID 12944331.
- ^ [není nutný primární zdroj ] Horowitz A, Shifman S, Rivlin N, Pisanté A, Darvasi A (2005). „Průzkum mikrodelece 22q11 u velké skupiny pacientů se schizofrenií“. Schizophr. Res. 73 (2–3): 263–7. doi:10.1016 / j.schres.2004.02.008. PMID 15653270.
- ^ Burn J (říjen 1999). „Uzávěrka pro CATCH22“. J. Med. Genet. 36 (10): 737–8. doi:10,1136 / jmg. 36.10.737. PMC 1734243. PMID 10528851.
- ^ A b Lindsay EA (listopad 2001). "Chromozomální mikrodelece: disekční syndrom del22q11". Nat. Genet. 2 (11): 858–68. doi:10.1038/35098574. PMID 11715041.
- ^ Swillen A, Vogels A, Devriendt K, Fryns JP (2000). „Syndrom delece chromozomu 22q11: aktualizace a hodnocení klinických rysů, kognitivně-behaviorálního spektra a psychiatrických komplikací“. Dopoledne. J. Med. Genet. 97 (2): 128–35. doi:10.1002 / 1096-8628 (200022) 97: 2 <128 :: AID-AJMG4> 3.0.CO; 2-Z. PMID 11180220.
- ^ Muldoon M, Ousley OY, Kobrynski LJ, Patel S, Oster ME, Fernandez-Carriba S, Cubells JF, Coleman K, Pearce BD (září 2015). „Účinek hypokalcemie v raném dětství na sociální a komunikační dovednosti související s autismem u pacientů se syndromem delece 22q11“. Eur Arch Psychiatry Clin Neurosci. 265 (6): 519–24. doi:10.1007 / s00406-014-0546-0. PMC 4379129. PMID 25267002.
- ^ Zinkstok J, van Amelsvoort T (2005). „Neuropsychologický profil a neuroimaging u pacientů se syndromem delece 22Q11.2: recenze“. Dítě Neuropsychol. 11 (1): 21–37. doi:10.1080/09297040590911194. PMID 15823981.
- ^ A b Butcher NJ, Kiehl TR, Hazrati LN, Chow EW, Rogaeva E, Lang AE, Bassett AS (2013). „Sdružení mezi časným nástupem Parkinsonovy choroby a delečním syndromem 22q11.2: identifikace nové genetické formy Parkinsonovy choroby a její klinické důsledky“. JAMA Neurol. 70 (11): 1359–66. doi:10.1001 / jamaneurol.2013.3646. PMC 4464823. PMID 24018986.
- ^ Mok KY, Sheerin U, Simón-Sánchez J, Salaka A, Chester L, Escott-Price V a kol. (Květen 2016). „Delece ve 22q11.2 u idiopatické Parkinsonovy nemoci: kombinovaná analýza dat asociace v celém genomu“. Lancet Neurol. 15 (6): 585–96. doi:10.1016 / S1474-4422 (16) 00071-5. PMC 4828586. PMID 27017469.
- ^ A b C D'Antonio LL, Scherer NJ, Miller LL, Kalbfleisch JH, Bartley JA (2001). "Analýza řečových charakteristik u dětí s velocardiofacial syndromem (VCFS) a dětí s fenotypovým překrytím bez VCFS". Rozštěp paláce Craniofac. J. 38 (5): 455–67. doi:10.1597 / 1545-1569 (2001) 038 <0455: AOSCIC> 2.0.CO; 2. ISSN 1545-1569. PMID 11522167.
- ^ A b C Scherer NJ, D'Antonio LL, Kalbfleisch JH (1999). „Časný vývoj řeči a jazyka u dětí s velokardiofaciálním syndromem“. Dopoledne. J. Med. Genet. 88 (6): 714–23. doi:10.1002 / (SICI) 1096-8628 (19991215) 88: 6 <714 :: AID-AJMG24> 3.0.CO; 2-B. PMID 10581495.
- ^ Scherer NJ, D'Antonio LL, Rodgers JR (2001). "Profily poruchy komunikace u dětí s velocardiofacial syndromem: srovnání s dětmi s Downovým syndromem". Genet. Med. 3 (1): 72–8. doi:10.1097/00125817-200101000-00016. PMID 11339384.
- ^ Eliez S, Palacio-Espasa F, Spira A (2000). „Malé děti se syndromem Velo-Cardio-Facial (CATCH-22). Psychologické a jazykové fenotypy“. Dětská psychiatrie Eur. 9 (2): 109–14. doi:10,1007 / s007870050005. PMID 10926060.
- ^ A b C Robin NH, Shprintzen RJ (2005). "Definování klinického spektra delece 22q11.2". J. Pediatr. 147 (1): 90–6. doi:10.1016 / j.jpeds.2005.03.007. PMID 16027702.
- ^ A b Solot CB, Knightly C, Handler SD (2000). "Poruchy komunikace u syndromu mikrodelece 22Q11.2". J Commun Disord. 33 (3): 187–203, kvíz 203–4. doi:10.1016 / S0021-9924 (00) 00018-6. PMID 10907715.
- ^ Persson C, Niklasson L, Oskarsdóttir S, Johansson S, Jönsson R, Söderpalm E (2006). „Jazykové znalosti u 5-8letých dětí se syndromem delece 22q11“. Int J Lang Commun Disord. 41 (3): 313–33. doi:10.1080/13682820500361497. PMID 16702096.
- ^ A b C Online Mendelian Inheritance in Man (OMIM): #188400
- ^ A b Packham EA, Brook JD (duben 2003). "Geny T-boxu u lidských poruch". Hučení. Mol. Genet. 12 Spec No 1 (90001): R37–44. doi:10,1093 / hmg / ddg077. PMID 12668595.
- ^ Tang KL, Antshel KM, Fremont WP, Kates WR (říjen 2015). „Behaviorální a psychiatrické fenotypy u syndromu delece 22q11.2“. J Dev Behav Pediatr. 36 (8): 639–50. doi:10.1097 / DBP.0000000000000210. PMC 4586411. PMID 26372046.
- ^ Maynard TM, Meechan DW, Dudevoir ML, Gopalakrishna D, Peters AZ, Heindel CC, Sugimoto TJ, Wu Y, Lieberman JA, Lamantia AS (listopad 2008). „Mitochondriální lokalizace a funkce podmnožiny kandidátních genů pro syndrom delece 22q11“. Mol. Buňka. Neurosci. 39 (3): 439–51. doi:10.1016 / j.mcn.2008.07.027. PMC 2729512. PMID 18775783.
- ^ A b Bartsch O, Nemecková M, Kocárek E, Wagner A, Puchmajerová A, Poppe M, Ounap K, Goetz P (únor 2003). "DiGeorge / velocardiofacial syndrom: FISH studie chromozomů 22q11 a 10p14 a klinické zprávy o proximální deleci 22q11". Dopoledne. J. Med. Genet. A. 117A (1): 1–5. doi:10,1002 / ajmg.a.10914. PMID 12548732.
- ^ Poirsier C, Besseau-Ayasse J, Schluth-Bolard C, Toutain J, Missirian C, Le Caignec C a kol. (Červen 2016). „Francouzská multicentrická studie s více než 700 pacienty s delecemi 22q11 diagnostikovanými pomocí FISH nebo aCGH“. Eur. J. Hum. Genet. 24 (6): 844–51. doi:10.1038 / ejhg.2015.219. PMC 4867458. PMID 26508576.
- ^ Edelmann L, Pandita RK, Spiteri E, Funke B, Goldberg R, Palanisamy N, Chaganti RS, Magenis E, Shprintzen RJ, Morrow BE (1999). „Společný molekulární základ pro poruchy přesmyku na chromozomu 22q11“. Hum Mol Genet. 8 (7): 1157–67. doi:10,1093 / hmg / 8.7.1157. PMID 10369860.
- ^ Jerome LA, Papaioannou VE (březen 2001). „Fenotyp DiGeorgeova syndromu u myší mutantních pro gen T-boxu, Tbx1“. Nat. Genet. 27 (3): 286–91. doi:10.1038/85845. PMID 11242110.
- ^ Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, Bradley A, Baldini A (březen 2001). „Tbx1 haploinsufficieny v oblasti DiGeorgeova syndromu způsobuje defekty aortálního oblouku u myší“. Příroda. 410 (6824): 97–101. doi:10.1038/35065105. PMID 11242049.
- ^ Chun S, Du F, Westmoreland JJ, Han SB, Wang YD, Eddins D a kol. (Leden 2017). „Thalamický miR-338-3p zprostředkovává sluchové thalamokortikální narušení a jeho pozdní nástup v modelech mikrodelece 22q11.2“. Nat. Med. 23 (1): 39–48. doi:10,1038 / nm. 4240. PMC 5218899. PMID 27892953.
- ^ „Gene (NCBI)“.
- ^ A b Kremer LS, Distelmaier F, Alhaddad B, Hempel M, Iuso A, Küpper C a kol. (2016). „Bialelické zkrácené mutace v TANGO2 způsobují opakované metabolické krize v počátečním věku s encefalokardiomyopatií“. American Journal of Human Genetics. 98 (2): 358–62. doi:10.1016 / j.ajhg.2015.12.009. PMC 4746337. PMID 26805782.
- ^ A b Lalani SR, Liu P, Rosenfeld JA, Watkin LB, Chiang T, Leduc MS a kol. (2016). „Recidivující svalová slabost s rhabdomyolýzou, metabolickými krizi a srdeční arytmií kvůli bialelickým mutacím TANGO2“. American Journal of Human Genetics. 98 (2): 347–57. doi:10.1016 / j.ajhg.2015.12.008. PMC 4746334. PMID 26805781.
- ^ Tonelli AR, Kosuri K, Wei S, Chick D (2007). „Záchvaty jako první projev syndromu delece chromozomu 22q11.2 u 40letého muže: kazuistika“. J Med Case Rep. 1: 167. doi:10.1186/1752-1947-1-167. PMC 2222674. PMID 18053182.
- ^ Miller, Kimberley A. (2008). "Diagnostika FISH syndromu vypuštění 22q11.2". Recenze ošetřovatelství novorozenců a kojenců. 8 (1): e11 – e19. doi:10.1053 / j.nainr.2007.12.006.
- ^ Jalali GR, Vorstman JA, Errami A, Vijzelaar R, Biegel J, Shaikh T, Emanuel BS (březen 2008). „Podrobná analýza 22q11.2 se sadou sond s vysokou hustotou MLPA“. Hučení. Mutat. 29 (3): 433–40. doi:10,1002 / humu.20640. PMC 2664158. PMID 18033723.
- ^ García-Herrero S, Campos-Galindo I, Martínez-Conejero JA, Serra V, Olmo I, Lara C, Simón C, Rubio C (2014). „Technologie BACs-on-Beads: spolehlivý test pro rychlou detekci aneuploidií a mikrodelecí v prenatální diagnostice“. Biomed Res Int. 2014: 590298. doi:10.1155/2014/590298. PMC 3985206. PMID 24795887.
- ^ Choy KW, Kwok YK, Cheng YK, Wong KM, Wong HK, Leung KO, Suen KW, Adler K, Wang CC, Lau TK, Schermer MJ, Lao TT, Leung TY (září 2014). „Diagnostická přesnost testu BACs-on-Beads ™ versus karyotypizace pro prenatální detekci chromozomálních abnormalit: retrospektivní řada po sobě jdoucích případů“. BJOG. 121 (10): 1245–52. doi:10.1111/1471-0528.12873. PMID 24893808.
- ^ Park SJ, Jung EH, Ryu RS, Kang HW, Ko JM, Kim HJ, Cheon CK, Hwang SH, Kang HY (květen 2011). „Klinická implementace celého genomu pole CGH jako testu první úrovně v 5080 před a po porodu“. Mol Cytogenet. 4: 12. doi:10.1186/1755-8166-4-12. PMC 3114015. PMID 21549014.
- ^ Mupanemunda, Richard H .; Watkinson, Michael (2004). Klíčová témata v neonatologii. CRC Press. str. 82. ISBN 9781859962343.
- ^ „DiGeorgeův syndrom (deleční syndrom 22q11.2)“. Klinika Mayo. Citováno 22. května 2020.
- ^ „DiGeorgeův syndrom (delece 22q11.2): Management a prognóza“. www.uptodate.com. Citováno 2018-10-30.
- ^ Markert ML, Devlin BH, Alexieff MJ, Li J, McCarthy EA, Gupton SE a kol. (Květen 2007). „Přehled 54 pacientů s úplnou anomálií DiGeorge zapsaných v protokolech pro transplantaci brzlíku: výsledek 44 po sobě jdoucích transplantací“. Krev. 109 (10): 4539–47. doi:10.1182 / krev-2006-10-048652. PMC 1885498. PMID 17284531.
- ^ „Klinická a metabolická genetika - klinika pro deleci 22q“. Nemocnice pro nemocné děti. Archivováno z původního dne 2016-04-07.
- ^ Fung WL, Butcher NJ, Costain G, Andrade DM, Boot E, Chow EW a kol. (Srpen 2015). „Praktické pokyny pro správu dospělých se syndromem delece 22q11.2“. Genet. Med. 17 (8): 599–609. doi:10.1038 / gim.2014.175. PMC 4526275. PMID 25569435.
- ^ Oskarsdóttir S, Vujic M, Fasth A (2004). „Incidence a prevalence syndromu delece 22q11: populační studie v západním Švédsku“. Oblouk. Dis. Dítě. 89 (2): 148–51. doi:10.1136 / adc.2003.026880. PMC 1719787. PMID 14736631.
- ^ Denně DK, Ardinger HH, Holmes GE (únor 2000). "Identifikace a hodnocení mentální retardace". Jsem známý lékař. 61 (4): 1059–67, 1070. PMID 10706158.
- ^ „Genetika 22q11.2 DS: demografie“. Informace pro lékaře. Klinika Dalglish Family Hearts and Minds pro dospělé s delečním syndromem 22q11.2. Archivováno z původního dne 9. března 2016. Citováno 26. srpna 2015.
- ^ „Di Georgeův syndrom“. Diagnostický kód 2015 ICD-10-CM D82.1. Archivováno z původního dne 24. září 2015. Citováno 26. srpna 2015.
- ^ A b "Velo-kardio-obličejový syndrom". Diagnostický kód 2015 ICD-10-CM Q93.81. Archivováno z původního dne 24. září 2015. Citováno 26. srpna 2015.
- ^ Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, Marino B, Oskarsdottir S, Philip N, Sullivan K, Swillen A, Vorstman J (srpen 2011). „Praktické pokyny pro správu pacientů se syndromem delece 22q11.2“. J. Pediatr. 159 (2): 332–9.e1. doi:10.1016 / j.jpeds.2011.02.039. PMC 3197829. PMID 21570089.
- ^ „Kampaň se stejným názvem - 22q.org“. 22q.org. Archivováno od originálu 10. 06. 2017. Citováno 2017-06-18.
Tento článek včlení public domain text z Americká národní lékařská knihovna
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |
- DiGeorgeův syndrom na Curlie
- McDonald-McGinn DM, Emanuel BS, Zackai EH (16. prosince 2005). „Syndrom mazání 22q11.2“. V publikacích Pagon RA, Bird TD, Dolan CR, Stephens K. (eds.). GeneReviews. PMID 20301696. NBK1523.
- Firth HV (17. února 2009). „22q11.2 Duplikace“. V publikacích Pagon RA, Bird TD, Dolan CR, Stephens K. (eds.). GeneReviews. PMID 20301749. NBK3823.
| Hlavní témata | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Přístupy | |||||||||||
| Práva, právo, podpora |
| ||||||||||
| Strukturální a pomocné | |||||||||||
| Sociální problémy | |||||||||||
| Umění, média, kultura, sport | |||||||||||
| |||||||||||