Kruhový chromozom 15 - Ring chromosome 15
| Kruhový chromozom 15 | |
|---|---|
| Ostatní jména | Syndrom kruhového chromozomu 15, kruh 15 |
 | |
| Vytvoření prstenu | |
| Specialita | Lékařská genetika |
| Doba trvání | celoživotní |
| Léčba | podpůrný |
| Frekvence | vzácný |
Kruhový chromozom 15 (někdy označováno jako r15) je stav, který vzniká, když chromozom 15 pojistky k vytvoření a prstencový chromozom. Obvykle se r15 tvoří v důsledku modifikací nebo delece genů na chromozomu 15 v předběžných stádiích embryonálního vývoje, ale zřídka jej lze také zdědit.[1]
Pokud se konce chromozomu spojí bez ztráty genetického materiálu, uchová si jedinec normální stav fenotyp s relativně malým rozdílem. Pokud však dojde k deleci genetické informace na distálních nestabilních koncích, kde dochází k fúzi subtelomerních struktur, vznikají syndromy spojené s daným konkrétním chromozomem.[2] Všechny chromozomy mají schopnost tvořit takové prstence; příznaky a závažnost závisí na množství ztracené genetické informace. Léčba r15 se tedy zaměřuje převážně na eradikaci těchto příznaků spíše než na samotný chromozomový kruh.
Prezentace

Jelikož počet hlášených případů je malý a fenotypová exprese syndromu kruhového chromozomu 15 se vyskytuje v širším spektru; buňky pacientů mohou mít různé úrovně mozaicismus.[3][4][5]
Kruhový chromozom 15 je spojen se zpožděním růstu v důsledku růstový faktor podobný inzulínu I odpor.[6]
Mezi běžné funkce patří zpoždění růstu, mentální retardace a vrozená vada.[7]
Z 25 studií zkoumaných Butlerem MG[3] všechny případy uváděly nedostatek růstu, 95% případů vykazovalo různé úrovně mentální retardace a 88% pacientů vykazovalo mikrocefalii. Kromě těch 3 hlavních příznaků, které se vyskytují nejčastěji, patří mezi další příznaky: Zpožděný kostní věk (7%) Hypertelorismus (46%), Brachydactyly (44%), Trojúhelníkový obličej (42%), Zpoždění řeči (39%), Čelní bossing (36%), anomální ucho (30%), Café-au-lait místa (30%), Kryptorchismus (30%) a srdeční abnormality (30%).[3]
Přesná, konkrétní asociace genotyp-fenotyp nebyla stanovena. Výzkum stavu se pokouší objasnit příčiny příznaků. Například přihlášením RYBA analýzy bylo zjištěno, že zpomalení růstu (nejběžnější fenotyp ) může být způsobeno terminální delecí 15q26 (specifická oblast na dlouhém rameni chromozomu 15).[8]
Mechanismus
Lidské tělo ukládá svůj genetický materiál do chromozomů. Počet chromozomy a genový lokus na chromozomu je pro každý druh jedinečný. Lidé mají 23 párů chromozomů, 22 párů autozomální chromozomy a 1 pár pohlavní chromozomy které rozlišují mezi muži a ženami.[9]Všechny lidské chromozomy mají 2 ramena - p (krátký) a q (dlouhé) rameno, které jsou odděleny centromery.[10]
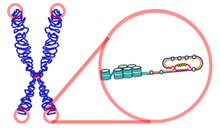
Existují dva navrhované mechanismy tvorby kruhového chromozomu. Jeden naznačuje, že chromozom 15 prochází před fúzí dvou zlomených ramen zkreslením obou paží a paží, což vede k velké ztrátě genetických materiálů. Pacienti tohoto typu r15 proto vykazují závažné klinické příznaky.[11]
Další navrhovaný mechanismus naznačuje přímé spojení dvou telomery bez ztráty kterékoli z telomerních a subtelomerních sekvencí. V důsledku toho je většina genetického materiálu zachována a příznaky jsou vyjádřeny v mírnější formě, což ztěžuje diagnostiku.[12]
Diagnóza

Ve většině případů se provádí postnatální diagnostika a do roku 2011 jsou prostřednictvím prenatální diagnostiky hlášeny pouze čtyři případy.[2]Vrozená brániční kýla a zpomalení nitroděložního růstu (tyto dva příznaky ohrožují pacienty rizikem r15) plodem ultrazvuk (Porodnická ultrasonografie ) v časovém období 16–24 týdnů je nutné provést další vyšetření a diagnostiku (jako je karyotypizace), aby se otestovala možnost kruhového chromozomu 15.[Citace je zapotřebí ]
Postnatální diagnóza
Pacienti by mohli být považováni za pacienty s prstencovým chromozomem 15, pokud se u nich zjistí: růstový nedostatek,[2] café-au-lait skvrny, zpoždění kostního věku a opičí záhyb nebo jiné dysmorfické rysy po narození.[Citace je zapotřebí ]
Řízení
Léčba onemocnění je založena na zmírnění příznaků.
Epidemiologie
Syndromy prstencových chromozomů jsou vzácné vrozené poruchy, které se pravděpodobně vyskytnou u mužů i žen, a příznaky lze pozorovat od narození, protože vznikají během embryonální fáze. Všechny rasy a etnické skupiny jsou náchylné k poruchám a riziko může být vyšší, pokud jsou rodiče nositeli, protože jsou geneticky zděděni.[Citace je zapotřebí ]
Lze zazvonit kterýkoli z 23 párů chromozomů a nedávná studie provedená „registrem lidských kruhových chromozomů“ v Číně odhalila, že častější hlášené formy kruhových chromozomů byly 13, 15, 18 a 22.[13]
Dějiny
r15 je neobvyklá genetická porucha, kterou poprvé zaznamenala doktorka Petrea Jacobsenová v roce 1966.[14] K dnešnímu dni bylo hlášeno přibližně 40 případů.[15]
Reference
- ^ "Prstencový chromozom 15 | Informační centrum o genetických a vzácných onemocněních (GARD) - program NCATS". rarediseases.info.nih.gov. Citováno 2019-04-11.
- ^ A b C Xu, F; Zou, CC; Liang, L; Huang, XM; Shao, YN (2011). „Ring Chromosome 15 Syndrome: Case Report and Literature Review“. HK J Paediatr (nová řada): 175–179.
- ^ A b C Butler, Merlin G .; Fogo, Agnes B .; Fuchs, David A .; Collins, Francis S .; Dev, Viathilingam G .; Phillips, John A .; Optiz, John M .; Reynolds, James F. (leden 1988). „Dva pacienti se syndromem kruhového chromozomu 15“. American Journal of Medical Genetics. 29 (1): 149–154. doi:10,1002 / ajmg.1320290119. ISSN 0148-7299. PMC 5083070. PMID 3278612.
- ^ Roback, Ellen W .; Barakat, Amin J .; Dev, V. G .; Mbikay, Majambu; Chrétien, Michel; Butler, Merlin G. (01.01.1991). „Dítě s delecí distálního dlouhého ramene chromozomu 15 (q26.1 → qter) a ztrátou genu receptoru pro růstový faktor 1 podobný inzulínu“. American Journal of Medical Genetics. 38 (1): 74–79. doi:10,1002 / ajmg.1320380117. ISSN 0148-7299. PMC 5493390. PMID 1849352.
- ^ Nuutinen, M; Kouvalainen, K; Knip, M (01.06.1995). „Dobrá růstová reakce na léčbu růstovým hormonem u syndromu kruhového chromozomu 15“. Journal of Medical Genetics. 32 (6): 486–487. doi:10,1136 / jmg.32.6.486. ISSN 1468-6244. PMC 1050492. PMID 7545237.
- ^ „Orphanet: Růst% 20odklad% 20due% 20to% 20inzulin jako% 20 růst% 20faktor% 20I% 20odpor“. www.orpha.net. Citováno 23. dubna 2020.
- ^ Peoples, R .; Milatovich, A .; Francke, U. (1995). „Hemizygosita v lokusu receptoru pro růstový faktor I podobného inzulínu (IGF1R) a selhání růstu u syndromu kruhového chromozomu 15“. Cytogenetický a genomový výzkum. 70 (3–4): 228–234. doi:10.1159/000134040. ISSN 1424-859X. PMID 7789178.
- ^ Harada, N; Shimokawa, O; Nagai, T; Kato, R; Kondoh, T; Niikawa, N; Matsumoto, N (08.10.2002). „Kritická oblast 4 Mb pro zpomalení intrauterinního růstu v 15q26“. Klinická genetika. 62 (4): 340–342. doi:10.1034 / j.1399-0004.2002.620416.x. ISSN 0009-9163. PMID 12372065.
- ^ Reference, Genetics Home. „Co je to chromozom?“. Genetická domácí reference. Citováno 2019-04-11.
- ^ „Definice dlouhého ramene chromozomu“. MedicineNet. Citováno 2019-04-11.
- ^ Kosztolanyi, G (únor 1987). „Existuje„ prstenový syndrom "? Analýza 207 kazuistik u pacientů s prstencovým autosomem." Genetika člověka. 75 (2): 174–179. doi:10.1007 / bf00591082. ISSN 0340-6717. PMID 3817812. S2CID 1596675.
- ^ Pezzolo, Annalisa; Gimelli, Giorgio; Cohen, Amnon; Lavaggetto, Antonella; Romano, Cesare; Fogu, Giuseppina; Zuffardi, Orsetta (srpen 1993). „Přítomnost telomerních a subtelomerních sekvencí ve fúzních bodech kruhových chromozomů naznačuje, že kruhový syndrom je způsoben nestabilitou kruhu“. Genetika člověka. 92 (1): 23–27. doi:10.1007 / bf00216140. ISSN 0340-6717. PMID 8365723. S2CID 23582495.
- ^ Hu, Qiping; Chai, Hongyan; Shu, Wei; Li, Peining (2018-02-27). „Registr chromozomů lidského kruhu pro případy v čínské populaci: opětovné zdůraznění cytogenomické a klinické heterogenity a přezkoumání diagnostických a léčebných strategií“. Molekulární cytogenetika. 11 (1): 19. doi:10.1186 / s13039-018-0367-3. ISSN 1755-8166. PMC 5828142. PMID 29492108.
- ^ JACOBSEN, PETREA (02.09.2009). „Prstencový chromozom ve skupině 13–15 spojený s mikrocefalickým nanismem, mentální retardací a emoční nezralostí“. Hereditas. 55 (2–3): 188–191. doi:10.1111 / j.1601-5223.1966.tb02047.x. ISSN 0018-0661.
- ^ Glass, Ian A .; Rauen, Katherine A .; Chen, Emily; Parkes, Jillian; Alberston, Donna G .; Pinkel, Daniel; Cotter, Philip D. (11.03.2005). Msgstr "Kruhový chromozom 15: charakterizace pomocí pole CGH". Genetika člověka. 118 (5): 611–617. doi:10.1007 / s00439-005-0030-z. ISSN 0340-6717. PMID 16267671. S2CID 24115238.