Seznam primárních imunodeficiencí - List of primary immunodeficiencies
Toto je seznam primární imunodeficience (PID), které jsou imunitní nedostatky které nejsou sekundární vůči jiné podmínce.
The Mezinárodní unie imunologických společností uznává devět tříd primárních imunodeficiencí, celkem přibližně 350 podmínek.[1] Aktualizace průvodce klasifikací z roku 2014 přidala 9. kategorii a přidala 30 nových genových defektů z předchozí verze 2009.[2][3] Nejnovější klasifikace byla vydána v roce 2017. Počet identifikovaných stavů v průběhu času stále roste, jak se provádí další výzkum.
Dopad primárních imunodeficiencí se pohybuje od mírného až po závažný na základě stavu.[4]
Kombinované imunodeficience T a B-buněk
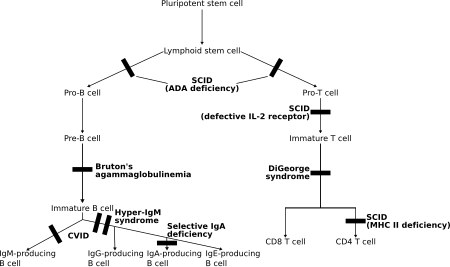
U těchto poruch obojí T lymfocyty a často B lymfocyty, regulátory adaptivní imunity, jsou nefunkční nebo jejich počet klesá. Hlavními členy jsou různé typy závažná kombinovaná imunodeficience (SCID).[5]
- T- / B + SCID (T buňky převážně chybí):
- nedostatek γc
- Nedostatek JAK3
- Receptor interleukinu-7 nedostatek
- CD45 nedostatek
- CD3δ, CD3ε nebo CD3ζ nedostatek
- Coronin-1A nedostatek
- LAT (gen) nedostatek
- T- / B- SCID (chybí T i B buňky)
- RAG 1/2 nedostatek
- DCLRE1C (Artemis) nedostatek
- XLF (protein) /Nedostatek Cernunnos
- DNA PKcs nedostatek
- DNA ligáza nedostatek typu IV
- adenosindeamináza (ADA) nedostatek
- retikulární dysgeneze
- Omennův syndrom
- Ligand CD40 nedostatek
- CD40 nedostatek
- CD3γ nedostatek
- CD8 nedostatek
- ICOS nedostatek
- ZAP70 nedostatek
- Nedostatek kanálu Ca ++
- MHC třída I nedostatek (s mutacemi v TAP1, TAP2, TAPBP nebo B2M )
- MHC třídy II nedostatek (s mutacemi v CIITA, RFXANK, RFX5 nebo RFXAP )
- Nedostatek CD25
- Nedostatek CD27
- Nedostatek STAT5b
- Nedostatek ITK
- Nedostatek SH2D1A (XLP1)
- Nedostatek MAGT1
- DOCK2 nedostatek
- DOCK8 nedostatek
- Nedostatek RhoH
- Aktivovaný delta syndrom PI3K
- MALT1 nedostatek
- BCL10 nedostatek
- BCL11B nedostatek
- KARTA 11 nedostatek
- Nedostatek MST1
- Nedostatek TCRα
- Nedostatek LCK
- IL-21 nedostatek
- Nedostatek IL-21R
- Nedostatek UNC119
- Nedostatek NIK
- Nedostatek OX40
- Nedostatek IKBKB
- TFRC nedostatek
- Moesin nedostatek
- RELB nedostatek
- Hypoplázie chrupavky
- Nedostatek LRBA
Převážně nedostatky protilátek
Primárně nedostatky protilátek, jeden nebo více izotypů imunoglobulin jsou sníženy nebo nefungují správně. Tyto proteiny, generované plazmatické buňky se obvykle váží na patogeny a zaměřují se na jejich zničení.[5]
- Chybějící B buňky s výslednou výraznou redukcí všech typů protilátek: X-vázaná agamaglobulinemie (Btk nedostatek, nebo Bruton agamaglobulinemie), μ -Těžký řetěz nedostatek, l 5 nedostatek, Igα nedostatek, BLNK nedostatek, thymom s imunodeficiencí
- B buňky nízké, ale přítomné nebo normální, ale se snížením 2 nebo více izotypů (obvykle IgG a IgA, někdy IgM): běžná variabilní imunodeficience (CVID), CD19 nedostatek, TACI Nedostatek (TNFRSF13B), BAFF receptor nedostatek.
- Normální počet B buněk se snížil IgG a IgA a zvýšil IgM: Hyper-IgM syndromy
- Normální počet B buněk s nedostatkem izotypu nebo lehkého řetězce: těžký řetěz vypuštění, řetěz kappa nedostatek, izolovaný nedostatek podtřídy IgG, IgA s nedostatkem podtřídy IgG, selektivní nedostatek imunoglobulinu A.
- Specifický nedostatek protilátek proti specifickým antigenům s normálními B buňkami a normálními koncentracemi Ig
- Přechodná dětská hypogamaglobulinémie (THI)
Další dobře definovaný syndrom imunodeficience
Řada syndromů uniká formální klasifikaci, ale jinak jsou rozpoznatelné podle konkrétních klinických nebo imunologických znaků.[5]
- Imunodeficience s trombocytopenií
- Oprava DNA vady nezpůsobující izolované SCID:
- Ataxia-telangiektázie
- Syndrom podobný ataxii
- Syndrom rozpadu Nijmegen
- Bloomův syndrom
- Syndrom imunodeficience - centromerická nestabilita - anomálie obličeje (ICF1, 2, 3 a 4)
- Nedostatek PMS2
- RIDDLE syndrom (Nedostatek RNF168)
- Nedostatek MCM4
- FILS syndrom (PÓL nedostatek)
- POLE2 nedostatek
- LIG1 nedostatek
- NSMCE3 nedostatek
- Nedostatek Hebo
- GINS1 nedostatek
- DiGeorgeův syndrom (pokud je spojen s brzlík vady)
- TBX1 nedostatek
- Syndrom CHARGE (CHD7 nedostatek nebo SEMA3E nedostatek)
- Okřídlená spirála /FOXN1 nedostatek
- Delece chromozomu 10p13-p14
- Imuno-kostní dysplázie (abnormální vývoj kostry s imunitními problémy):
- Hypoplázie chrupavky a vlasů
- Schimkeho syndrom
- MYSM1 nedostatek
- MOPD1 nedostatek
- EXTL3 nedostatek
- Hyper IgE syndromy
- Jobův syndrom (STAT3 nedostatek)
- Comel-Nethertonův syndrom
- PGM3 nedostatek
- Hypohidrotická ektodermální dysplázie
- Poruchy vápníkového kanálu
- ORAI1 nedostatek
- Nedostatek STIM1
- Transkobalamin 2 nedostatek
- Imunodeficience s mnohočetnými atresiemi střeva (TTC7A nedostatek)
- Venookluzivní onemocnění jater s imunodeficiencí (VODI)
- Viciho syndrom
- Nedostatek purinové nukleosidové fosforylázy (PNP)
- AR-DKC (vrozená autosomálně dominantní dyskeratóza)
- Hermanského – Pudlakův syndrom typ 2
- Chronická mukokutánní kandidóza
- HOIL1 nedostatek
- HOIP nedostatek
- XL-dyskeratosis congenita (Hoyeraal-Hreidarssonův syndrom )
- Syndrom Hennekam lymfangiektázie-lymfedém
- Kabukiho syndrom
- Nedostatek MTHFD1
- Nedostatek STAT5b
- Nedostatek IKAROS
Nemoci imunitní dysregulace
Za určitých podmínek je převládajícím problémem spíše regulace než vnitřní aktivita částí imunitního systému.[5]
- Imunodeficience s hypopigmentací nebo albinismus: Syndrom Chédiak – Higashi, Griscelliho syndrom typ 2
- Familiární hemofagocytární lymfohistiocytóza: perforin nedostatek, UNC13D nedostatek, syntaxin 11 nedostatek
- X-vázaný lymfoproliferativní syndrom
- Syndromy s autoimunitou:
- (A) Autoimunitní lymfoproliferativní syndrom: typ 1a (CD95 vady), typ 1b (Fas ligand závady), typ 2a (CASP10 vady), typ 2b (CASP8 vady)
- b) OČEKÁVÁNO (autoimunitní polyendokrinopatie s kandidózou a ektodermální dystrofií)
- (C) IPEX (imunodysregulační polyendokrinopatie, enteropatie, X-vázaný syndrom)
- d) Nedostatek CD25
Vrozené vady počtu, funkce nebo obou fagocytů
Fagocyty jsou buňky, které pohlcují a přijímají patogeny (fagocytóza ) a zničte je chemickými látkami. Monocyty /makrofágy stejně jako granulocyty jsou schopni tohoto procesu. Za určitých podmínek je buď snížen počet fagocytů, nebo je narušena jejich funkční kapacita.[5]
- Těžká vrozená neutropenie: kvůli ELA2 nedostatek (s myelodysplasie )
- Těžká vrozená neutropenie: kvůli GFI1 nedostatek (s T / B lymfopenií)
- Elastáza nedostatek
- Kostmann syndrom (HAX1 nedostatek)
- Neutropenie se srdečními a urogenitálními malformacemi
- Glykogenová skladovací choroba typu 1b
- Cohenův syndrom
- Clericuzio syndrom
- Cyklická neutropenie
- X-vázaná neutropenie / myelodysplasie
- Nedostatek P14
- HYOU1 nedostatek
- JAGN1 nedostatek
- SMARCD2 nedostatek
- 3-methylglutakonová acidurie
- Deficit adheze leukocytů typu 1
- Nedostatek adheze leukocytů typ 2
- Nedostatek adheze leukocytů typ 3
- RAC2 nedostatek (Syndrom imunodeficience neutrofilů )
- Beta-aktin nedostatek
- Receptor G-CSF nedostatek
- Lokalizovaná juvenilní paradentóza
- Papillon – Lefèvrov syndrom
- Specifický nedostatek granulí
- Shwachman-Diamondův syndrom
- WDR1 nedostatek
- Cystická fibróza
- Chronické granulomatózní onemocnění: X-vázané nebo autosomální (CYBA, NCF1, NCF2, NCF4 )
- IL-12 a IL-23 Nedostatek β1 řetězce
- IL-12p40 nedostatek
- Nedostatek glukóza-6-fosfát dehydrogenázy třída 1
- Interferonový y receptor 1 nedostatek
- Interferonový y receptor 2 nedostatek
- STAT1 nedostatek
- MKL1 nedostatek
- INZERÁT hyper-IgE
- AR hyper-IgE
- Plicní alveolární proteinóza
- MonoMac syndrom (Nedostatek GATA2 )
Poruchy vrozené imunity
Několik vzácných stavů je způsobeno vadami v vrozený imunitní systém, což je základní obranná linie nezávislá na pokročilejších systémech souvisejících s lymfocyty. Mnoho z těchto stavů je spojeno s kožními problémy.[5]
- Receptor interleukinu 12, beta 1 nedostatek
- IL-12p40 nedostatek
- Interferonový gama receptor 1 nedostatek
- Interferonový gama receptor 2 nedostatek
- Nedostatek Tyk2
- JAK1 ztráta funkce
- ISG15 nedostatek
- RORc nedostatek
- STAT1 nedostatek, mutace zesílení funkce
- STAT2 nedostatek
- IRF7 nedostatek
- CD16 nedostatek
- IRF8 nedostatek
- IFNAR2 nedostatek
- Nedostatky dráhy TLR
- MDA5 nedostatek
- Epidermodysplasia verruciformis
- WHIM syndrom (bradavice, hypogamaglobulinémie, infekce, myelokathexis)
- EVER1 a EVER2 nedostatek
- Herpes simplex encefalitida
- KARTA 9 nedostatek
- Chronická mukokutánní kandidóza
- Trypanosomiáza
- RPSA nedostatek s vrozená asplenie
- HMOX nedostatek s vrozenou asplenií
- CLCN7 nedostatek s osteoporóza
- OSTM1 nedostatek s osteoporózou
- Hidradenitis suppurativa
Autozánětlivé poruchy
Spíše než předisponovat k infekcím vede většina autoinflamačních poruch k nadměrnému zánětu. Mnoho se projevuje jako syndromy periodické horečky. Mohou přímo zahrnovat různé orgány, stejně jako předisponovat k dlouhodobému poškození (např. Tím, že vedou k amyloid depozice).[5]
- Rodinná středomořská horečka
- Aicardi – Goutièresův syndrom s TREX1, SAMHD1 nebo IFIH1 mutace
- Spondyloenchondro-dysplázie s imunitní dysregulací (ACP5 mutace)
- Vazculopatie spojená se STING s nástupem v kojeneckém věku
- Síťovaná pigmentová porucha vázaná na X
- USP18 nedostatek
- SVÍČKA (Chronická atypická neutrofilní dermatitida s lipodystrofií)
- Singleton-Mertenův syndrom
- Periodický syndrom spojený s TNF receptorem (DOPRAVY)
- Hyper-IgD syndrom (Nedostatek mevalonátkinázy )
- CIAS1 související nemoci:
- NLRP1 nedostatek
- Syndrom PAPA (pyogenní sterilní artritida, pyoderma gangrenosum, akné)
- 17. ADAM nedostatek
- Blauův syndrom
- Majeedův syndrom (Chronická rekurentní multifokální osteomyelitida a vrozená dyserytropoetická anémie)
- DIRA (nedostatek antagonisty receptoru IL-1)
- DITRA (nedostatek antagonisty receptoru IL-36)
- CARD14 zprostředkovaná psoriáza (TÁBORY)
- Cherubismus
- Vada COPA
- Otulipenia / ORAS
Doplňte nedostatky
The doplňkový systém je součástí vrozeného i adaptivního imunitního systému; je to skupina cirkulujících proteinů, které mohou vázat patogeny a tvořit membránový atakující komplex. Doplňte nedostatky jsou výsledkem nedostatku některého z těchto proteinů. Mohou mít predispozici k infekcím, ale také k autoimunitním podmínkám.[5]
- Nedostatek C1q (syndrom podobný lupusu, revmatoidní onemocnění, infekce)
- Nedostatek C1r (idem)
- Nedostatek C1s
- Nedostatek C4 (syndrom podobný lupusu)
- Nedostatek C2 (syndrom podobný lupusu, vaskulitida, polymyositida, pyogenní infekce )
- Nedostatek C3 (opakující se pyogenní infekce )
- Nedostatek C5 (Neisseriální infekce, SLE)
- Nedostatek C6 (idem)
- Nedostatek C7 (idem, vaskulitida)
- Nedostatek C8a
- Nedostatek C8b
- Nedostatek C9 (Neisseriální infekce)
- Nedostatek inhibitoru C1 (dědičný angioedém)
- Nedostatek faktoru I. (pyogenní infekce )
- Nedostatek faktoru H. (hemolyticko-uremický syndrom, membranoproliferativní glomerulonefritida )
- Nedostatek faktoru D. (Neisseriální infekce)
- Nedostatek správnéhodin (Neisseriální infekce)
- Nedostatek MBP (pyogenní infekce )
- Nedostatek MASP2
- Doplňte nedostatek receptoru 3
- Nedostatek membránového kofaktorového proteinu (CD46)
- Nedostatek inhibitoru komplexu membránového záchvatu (CD59)
- Paroxysmální noční hemoglobinurie
- Nedostatek Ficolin 3
- Nedostatek správnéhodin
- Nedostatek faktoru I.
- Nedostatek faktoru H.
- Nedostatek trombomodulinu
- CHAPEL nemoc
Fenokopie primárních imunitních nedostatků
- Autoimunitní lymfoproliferativní syndrom
- Autoimunitní leukoproliferativní porucha spojená s RAS
- Velká granulovaná lymfocytóza
- Atypický hemolyticko-uremický syndrom
- Dobrý syndrom
Reference
- ^ Bousfiha, Aziz; Jeddane, Leila; Picard, Capucine; Ailal, Fatima; Bobby Gaspar, H .; Al-Herz, Waleed; Chatila, Talal; Crow, Yanick J. (2018). „Fenotypová klasifikace IUIS 2017 pro primární imunodeficience“. Journal of Clinical Immunology. 38 (1): 129–143. doi:10.1007 / s10875-017-0465-8. ISSN 0271-9142. PMC 5742599. PMID 29226301.
- ^ Waleed Al-Herz; Aziz Bousfiha; Jean-Laurent Casanova; et al. (2014). „Nemoci primární imunodeficience: aktualizace klasifikace z Odborného výboru Mezinárodní unie pro imunologické společnosti pro primární imunodeficienci“ (PDF). Hranice v imunologii. 5 (162): 1–33. doi:10.3389 / fimmu.2014.00162. PMC 4001072. PMID 24795713.
- ^ Notarangelo L, Casanova JL, Conley ME a kol. (2006). „Nemoci primární imunodeficience: aktualizace ze zasedání Výboru pro klasifikaci nemocí primární imunodeficience v Budapešti, 2005“. J. Allergy Clin. Immunol. 117 (4): 883–96. doi:10.1016 / j.jaci.2005.12.1347. PMID 16680902.
- ^ „Společný variabilní imunitní deficit“. NORD (Národní organizace pro vzácné poruchy). Citováno 5. března 2019.
- ^ A b C d E F G h Notarangelo LD, Fischer A, Geha RS a kol. (Prosinec 2009). „Primární imunodeficience: aktualizace z roku 2009: Výbor odborníků na primární imunodeficience (PID) Mezinárodní unie imunologických společností (IUIS)“. J. Allergy Clin. Immunol. 124 (6): 1161–78. doi:10.1016 / j.jaci.2009.10.013. PMC 2797319. PMID 20004777.