Trombotická trombocytopenická purpura - Thrombotic thrombocytopenic purpura
| Trombotická trombocytopenická purpura | |
|---|---|
| Ostatní jména | Moschcowitzův syndrom,[1] idiopatická trombotická trombocytopenická purpura[2] |
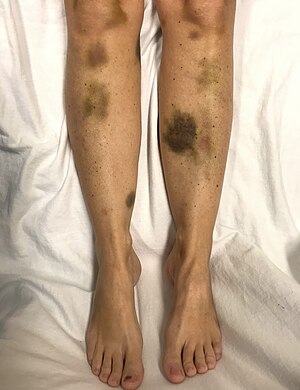 | |
| Spontánní modřiny u ženy s kriticky nízkými krevními destičkami | |
| Specialita | Hematologie |
| Příznaky | Velké modřiny, horečka slabost, dušnost zmatek, bolest hlavy[3][2] |
| Obvyklý nástup | Dospělost[3] |
| Příčiny | Neznámý, bakteriální infekce určité léky, autoimunitní onemocnění, těhotenství[3] |
| Diagnostická metoda | Na základě příznaků a krevních testů[2] |
| Diferenciální diagnostika | Hemolyticko-uremický syndrom (HUS), atypický hemolyticko-uremický syndrom (aHUS)[4] |
| Léčba | Výměna plazmy, imunosupresiva[1] |
| Prognóza | <20% riziko úmrtí[1] |
| Frekvence | 1 ze 100 000 lidí[3] |
Trombotická trombocytopenická purpura (TTP) je krevní porucha což má za následek krevní sraženiny formování v malém cévy v celém těle.[2] Výsledkem je a nízký počet krevních destiček, nízký počet červených krvinek v důsledku jejich rozpadu a často ledviny, srdce, a mozek dysfunkce.[1] Příznaky mohou zahrnovat velké modřiny, horečka slabost, dušnost zmatek a bolest hlavy.[2][3] Mohou se objevit opakované epizody.[3]
V přibližně polovině případů je identifikován spouštěč, zatímco ve zbývající části zůstává příčina neznámá.[3] Mezi známé spouštěče patří bakteriální infekce určité léky, autoimunitní onemocnění jako lupus, a těhotenství.[3] Základní mechanismus obvykle zahrnuje protilátky inhibování enzym 13. ADAMTS.[1] To má za následek snížené členění velkých multimery z von Willebrandův faktor (vWF) na menší jednotky.[1] Méně často je TTP zdědil od rodičů člověka, známý jako Upshaw – Schulmanův syndrom taková, že dysfunkce ADAMTS13 je přítomna od narození.[5] Diagnóza je obvykle založena na příznacích a krevních testech.[2] To může být podpořeno měřením aktivity nebo protilátek proti ADAMTS13.[2]
S výměna plazmy riziko úmrtí se snížilo z více než 90% na méně než 20%.[1] Imunosupresiva, jako glukokortikoidy, a rituximab lze také použít.[3] Transfuze krevních destiček se obecně nedoporučují.[6]
Postiženo je přibližně 1 na 100 000 lidí.[3] Nástup je obvykle v dospělosti a ženy jsou častěji postiženy.[3] Asi 10% případů začíná v dětství.[3] Tento stav poprvé popsal Eli Moschcowitz v roce 1924.[3] Základní mechanismus byl určen v 80. a 90. letech.[3]
Příznaky a symptomy
Známky a příznaky TTP mohou být nejprve jemné a nespecifické. Mnoho lidí zažívá chřipkové nebo průjmové onemocnění před rozvojem TTP.[7] Neurologické příznaky jsou velmi časté a jejich závažnost se velmi liší. Mezi často uváděné příznaky patří cítit se velmi unavený, zmatek, a bolesti hlavy.[7] Záchvaty a příznaky podobné těm z a mrtvice lze také vidět.[7] Mezi další příznaky mimo jiné patří žloutenka nebo bledost kůže, rychlý srdeční rytmus nebo dušnost nebo přesné fialové nebo načervenalé tečky na kůži známé jako petechie.[Citace je zapotřebí ]
Jak TTP postupuje, tvoří se krevní sraženiny v malých krevních cévách (mikrovaskulatura) a destičky (buňky srážení) se spotřebovávají. Výsledkem může být modřina a vzácně krvácení. Podlitiny mají často podobu purpura, zatímco nejčastějším místem krvácení, pokud k němu dojde, je z nosu nebo dásní. Větší modřiny (ekchymózy ) se také může vyvinout.[Citace je zapotřebí ]Klasická prezentace TTP, která se vyskytuje u méně než 10% lidí, zahrnuje pět lékařských příznaků.[3] Tyto jsou:
- Horečka
- Změny duševního stavu
- Trombocytopenie
- Snížená funkce ledvin
- Hemolytická anémie (mikroangiopatická hemolytická anémie ).[7]
Vysoký krevní tlak (hypertenze ) lze nalézt při vyšetření.[8]
Příčiny
TTP, stejně jako u jiných mikroangiopatických hemolytických anémií (MAHA), je způsoben spontánně agregace destiček a aktivace koagulace v malých cévách. Trombocyty se spotřebovávají v procesu agregace a vážou vWF. Tyto komplexy destička-vWF tvoří malé krevní sraženiny, které cirkulují v cévách a způsobují stříhání červených krvinek, což vede k jejich prasknutí a formování schistocyty.[9]Dvě nejlépe pochopené příčiny TTP jsou způsobeny autoimunitou a dědičným nedostatkem přípravku ADAMTS13 (známý jako Upshaw-Schülmanův syndrom).[9] Většina zbývajících případů je sekundární vůči nějakému jinému faktoru.[Citace je zapotřebí ]
Autoimunitní
TTP neznámé příčiny byl dlouho známý jako idiopatické TTP, ale v roce 1998 se ukázalo, že většina případů byla způsobena inhibicí enzymu 13. ADAMTS podle protilátky. Vztah redukovaného 13. ADAMTS k patogenezi TTP je známá jako Furlan-Tsai hypotéza, po dvou nezávislých skupinách výzkumníků, kteří publikovali svůj výzkum ve stejném čísle New England Journal of Medicine.[10][11][12] Tyto případy jsou nyní klasifikovány jako autoimunitní onemocnění a jsou známé jako autoimunitní TTP (nezaměňovat s imunitní / idiopatická trombocytopenická purpura ).[Citace je zapotřebí ]
ADAMTS13 je a metaloproteináza odpovědný za rozdělení von Willebrandův faktor (vWF), protein, který spojuje krevní destičky, krevní sraženiny a stěna krevních cév v procesu srážení krve. Velmi velké multimery vWF jsou náchylnější k koagulaci. Bez řádného štěpení vWF pomocí ADAMTS13 tedy dochází ke koagulaci vyšší rychlostí, zejména v mikrovaskulatuře, části systému krevních cév, kde je vWF nejaktivnější kvůli vysokému smykové napětí.[5] U idiopatické TTP lze u většiny (80%) lidí detekovat výrazně sníženou (<5% normální) aktivitu ADAMTS13 a v této podskupině se často nacházejí inhibitory (44–56%).
Genetický

Tento stav může být také vrozený. Takové případy mohou být způsobeny mutacemi genu ADAMTS13.[15] Tato dědičná forma TTP se nazývá Upshaw – Schulmanův syndrom.[16][17][18] Lidé s tímto zděděným nedostatkem ADAMTS13 mají překvapivě mírný fenotyp, ale TTP se u nich vyvine v klinických situacích se zvýšenými hladinami von Willebrandova faktoru, např. infekce. Údajně méně než 1% všech případů TTP je způsobeno syndromem Upshaw – Schulman.[19] Lidé s tímto syndromem mají obvykle 5–10% normální aktivity ADAMTS-13.[18][20]
Sekundární
Sekundární TTP je diagnostikována, když historie osoby zmiňuje jeden ze známých rysů spojených s TTP. Zahrnuje asi 40% všech případů TTP. Predisponujícími faktory jsou:[9]
- Rakovina
- Transplantace kostní dřeně
- Těhotenství
- Léky použití:
- Antivirotika (acyklovir )
- Určitý chemoterapie léky jako gemcitabin a mitomycin C.
- Chinin
- Oxymorfon
- Kvetiapin
- Bevacizumab
- Sunitinib
- Inhibitory agregace krevních destiček (tiklopidin, klopidogrel, a prasugrel )
- Imunosupresiva (cyklosporin, mitomycin, takrolimus / FK506, interferon-α )
- Léky ovlivňující hormony (estrogeny, antikoncepce, hormonální substituční léčba)[21]
- HIV-1 infekce
Mechanismus sekundárního TTP je špatně pochopen, protože aktivita ADAMTS13 obecně není tak depresivní jako u idiopatického TTP a inhibitory nelze detekovat. Pravděpodobná etiologie může zahrnovat, alespoň v některých případech, poškození endotelu,[22] ačkoli tvorba trombů vedoucí k okluzi cév nemusí být v patogenezi sekundárního TTP nezbytná.[23] Tyto faktory lze také považovat za formu sekundárního aHUS; lidé s těmito vlastnostmi jsou proto potenciálními kandidáty na protikomplementovou terapii.
Mechanismus
Základní mechanismus obvykle zahrnuje autoprotilátkami zprostředkovanou inhibici enzym 13. ADAMTS, a metaloproteáza zodpovědný za štěpení velkých multimerů von Willebrandův faktor (vWF) na menší jednotky. Zvýšení cirkulujících multimerů vWF zvyšuje adhezi krevních destiček k oblastem endoteliální zranění, zvláště kde arterioly a kapiláry setkat, což má za následek tvorbu malých krevních sraženin nazývaných tromby. Protože se krevní destičky vyčerpávají při tvorbě trombů, vede to ke snížení počtu celkově cirkulujících krevních destiček, což může způsobit život ohrožující krvácení. Červené krvinky procházející mikroskopickými sraženinami jsou vystaveny smykové napětí, což poškozuje jejich membrány, což vede k prasknutí červených krvinek v krevních cévách, což zase vede k anémie a schistocyt formace. Přítomnost těchto krevní sraženiny v malých cévách snižuje průtok krve do orgánů, což vede k poškození buněk a poškození koncových orgánů.[Citace je zapotřebí ]
Diagnóza
Diferenciální diagnostika
TTP se vyznačuje trombotická mikroangiopatie (TMA), tvorba krevních sraženin v malých krevních cévách v celém těle, což může vést k mikroangiopatické hemolytické anémii a trombocytopenii. Tuto vlastnost sdílejí dva příbuzné syndromy, hemolyticko-uremický syndrom (HUS) a atypický hemolyticko-uremický syndrom (aHUS).[4] Proto je nezbytná diferenciální diagnostika těchto onemocnění způsobujících TMA. Kromě TMA může být u každého z těchto onemocnění přítomen jeden nebo více z následujících příznaků: neurologické příznaky (např. Zmatenost,[24][25] mozkové křeče[25] záchvaty,[26]); poškození ledvin[27] (např. zvýšené kreatinin,[28] snížená odhadovaná rychlost glomerulární filtrace [eGFR],[28] abnormální analýza moči[29]); a gastrointestinální (GI) příznaky (např. průjem[24][30] nevolnost / zvracení,[26] bolest břicha,[26] gastroenteritida.[24][27] Na rozdíl od HUS a aHUS je známo, že TTP je způsoben získaným defektem proteinu ADAMTS13, takže laboratorní test ukazující ≤ 5% normálních hladin ADAMTS13 naznačuje TTP.[31] Hladiny ADAMTS13 nad 5% spolu s pozitivním testem na shiga-toxin / enterohemoragický E-coli (EHEC), spíše svědčí o HUS,[32] zatímco absence shiga-toxinu / EHEC může potvrdit diagnózu aHUS.[31]
Léčba
Vzhledem k vysoké úmrtnosti neléčeného TTP, a předpokládaná diagnóza TTP se tvoří, i když je vidět pouze mikroangiopatická hemolytická anémie a trombocytopenie, a je zahájena léčba. Transfúze je u trombotického TTP kontraindikována, protože podporuje koagulopatii. Od počátku 90. let plazmaferéza se stalo léčbou volby pro TTP.[33][34] Tohle je výměna transfuze zahrnující odebrání osoby krevní plazma přes aferéza a nahrazení dárcovskou plazmou (čerstvá zmrazená plazma nebo kryosupernatant ); postup je nutné opakovat denně, aby se vyloučil inhibitor a zmírnily příznaky. Pokud není k dispozici aferéza, může být nalita čerstvá zmrazená plazma, ale objem, který lze bezpečně podat, je omezen kvůli nebezpečí přetížení tekutinami.[35] Samotná plazmatická infuze není tak prospěšná jako výměna plazmy.[33] Kortikosteroidy (prednison nebo prednisolon ) jsou obvykle uvedeny.[34] Rituximab, a monoklonální protilátka zaměřené na CD20 molekula na B lymfocyty, lze použít při diagnostice; předpokládá se, že to zabíjí B buňky a tím snižuje produkci inhibitoru.[34] Silnější doporučení pro rituximab existuje tam, kde TTP nereaguje na kortikosteroidy a plazmaferézu.[34]
Kaplacizumab je alternativní možností při léčbě TTP, protože se ukázalo, že ve srovnání s lidmi, kteří užívali placebo, vyvolává rychlejší vymizení nemoci.[36] Použití kaplacizumabu však bylo u studovaných subjektů spojeno se zvýšenou tendencí ke krvácení.[Citace je zapotřebí ]
Lidé s refrakterní nebo relabující TTP mohou obdržet další imunosupresivní terapie, např. vinkristin, cyklofosfamid, cyklosporin A nebo splenektomie.[3][35]
Děti se syndromem Upshaw-Schülman dostávají profylaktickou plazmu každé dva až tři týdny; to udržuje přiměřenou úroveň fungování ADAMTS13. Někteří tolerují delší intervaly mezi infuzemi plazmy. K vyvolání událostí, jako je chirurgický zákrok, mohou být nutné další infuze plazmy; alternativně může být počet krevních destiček pečlivě sledován kolem těchto příhod při podávání plazmy, pokud počet poklesne.[37]
Měření krevních hladin laktátdehydrogenáza, krevní destičky a schistocyty se používají k monitorování progrese onemocnění nebo remise.[Citace je zapotřebí ] Aktivitu a hladiny inhibitoru ADAMTS13 lze měřit během sledování, ale u pacientů bez příznaků se použití rituximabu nedoporučuje.[34]
Prognóza
Míra úmrtnosti se u neléčených případů pohybuje kolem 95%, ale prognóza je přiměřeně příznivá (80–90% přežití) u lidí s idiopatickou TTP diagnostikovanou a léčenou včas plazmaferéza.[38]
Epidemiologie
Výskyt TTP je přibližně 4–5 případů na milion lidí ročně.[39] Idiopatická TTP se vyskytuje častěji u žen a lidí afrického původu a TTP sekundární k autoimunitním poruchám, jako je systémový lupus erythematodes vyskytuje se častěji u lidí afrického původu, ačkoli jiné sekundární formy toto rozdělení nevykazují.[40] Těhotné ženy a ženy v EU post partum období představovalo významnou část (12–31%) případů v některých studiích; TTP postihuje přibližně jedno z 25 000 těhotenství.[41]
Dějiny
TTP původně popsal Eli Moschcowitz na Nemocnice Beth Israel v New York City v roce 1925. Moschcowitz připisoval nemoc (nesprávně, jak je nyní známo) toxické příčině. Moschcowitz zaznamenal, že jeho pacientka, 16letá dívka, měla anémii, malý a velké modřiny, mikroskopické hematurie, a při pitvě, šířeny mikrovaskulární tromby.[42] V roce 1966 vedl přehled 16 nových případů a 255 dříve hlášených případů k formulaci klasické pentády symptomů a nálezů (tj. Trombocytopenie, mikroangiopatická hemolytická anémie, neurologické příznaky, selhání ledvin, horečka); v této sérii bylo zjištěno, že úmrtnost je velmi vysoká (90%).[43]
Zatímco reakce na transfuzi krve byla zaznamenána dříve, zpráva z roku 1978 a následné studie ukázaly, že krevní plazma byla vysoce účinná při zlepšování procesu onemocnění.[44] V roce 1991 se uvádí, že výměna plazmy poskytuje lepší míru odezvy ve srovnání s infuzí plazmy.[45]V roce 1982 byla nemoc spojena s abnormálně velkými multimery von Willebrandova faktoru. Identifikace deficitu proteáza u lidí s TTP bylo vyrobeno v roce 1998. Umístění ADAMTS13 v lidském genomu bylo identifikováno v roce 2001.[44]
Reference
- ^ A b C d E F G Kremer Hovinga, JA; Coppo, P; Lämmle, B; Moake, JL; Miyata, T; Vanhoorelbeke, K (6. dubna 2017). "Trombotická trombocytopenická purpura". Recenze přírody. Primery nemoci. 3: 17020. doi:10.1038 / nrdp.2017.20. PMID 28382967. S2CID 11960153.
- ^ A b C d E F G "Trombotická trombocytopenická purpura, získaná". Informační centrum o genetických a vzácných onemocněních (GARD) - program NCATS. Citováno 10. října 2018.
- ^ A b C d E F G h i j k l m n Ó p Joly, BS; Coppo, P; Veyradier, A (25. května 2017). „Trombotická trombocytopenická purpura“. Krev. 129 (21): 2836–2846. doi:10.1182 / krev-2016-10-709857. PMID 28416507.
- ^ A b George JN (listopad 2010). „Jak zacházím s pacienty s trombotickou trombocytopenickou purpurou: 2010“. Krev. 116 (20): 4060–9. doi:10.1182 / krev-2010-07-271445. PMID 20686117.
- ^ A b Moake JL (2004). „von Willebrandův faktor, ADAMTS-13 a trombotická trombocytopenická purpura“. Semin. Hematol. 41 (1): 4–14. doi:10.1053 / j.seminhematol.2003.10.003. PMID 14727254.
- ^ Wood, Marie E .; Philips, George K. (2003). Hematologická / onkologická tajemství. Elsevier Health Sciences. p. 68. ISBN 978-1560535164.
- ^ A b C d Shatzel, JJ; Taylor, JA (březen 2017). "Syndromy trombotické mikroangiopatie". Lékařské kliniky Severní Ameriky (Posouzení). 101 (2): 395–415. doi:10.1016 / j.mcna.2016.09.010. PMID 28189178.
- ^ Allford S, Machin S (2005). „Trombotická trombocytopenická purpura“. NetDoctor.co.uk.
- ^ A b C Moake JL (2002). "Trombotické mikroangiopatie". N. Engl. J. Med. 347 (8): 589–600. doi:10.1056 / NEJMra020528. PMID 12192020.
- ^ Moake JL (1998). "Moschcowitz, multimery a metaloproteáza". N. Engl. J. Med. 339 (22): 1629–31. doi:10.1056 / NEJM199811263392210. PMID 9828253.
- ^ Furlan M, Robles R, Galbusera M a kol. (1998). „proteáza štěpící faktor von Willebranda při trombotické trombocytopenické purpuře a hemolyticko-uremickém syndromu“. N. Engl. J. Med. 339 (22): 1578–84. doi:10.1056 / NEJM199811263392202. PMID 9828245.
- ^ Tsai HM, Lian EC (1998). „Protilátky proti von Willebrandovu faktoru štěpícímu proteázu při akutní trombotické trombocytopenické purpuře“. N. Engl. J. Med. 339 (22): 1585–94. doi:10.1056 / NEJM199811263392203. PMC 3159001. PMID 9828246.
- ^ „OMIM Entry - # 274150 - TROMBOTICKÁ TROMBOCYTOPENICKÁ PURPURA, KONGENITÁLNÍ; TTP“. www.omim.org. Citováno 23. října 2017.
- ^ VYHRAZENO, VLOŽTE NÁS 14 - VŠECHNA PRÁVA. „Orphanet: Trombotická trombocytopenická purpura“. www.orpha.net. Citováno 23. října 2017.
- ^ Conboy E, Partain PI, Warad D, Kluge ML, Arndt C, Chen D, Rodriguez V (2017) Závažný případ vrozené trombotické trombocytopenie purpury vyplývající ze složené heterozygosity zahrnující novou patogenní variantu ADAMTS13. J Pediatr Hematol Oncol
- ^ Schulman I, Pierce M, Lukens A, Currimbhoy Z (červenec 1960). „Studie trombopoézy. I. Faktor v normální lidské plazmě vyžadovaný pro produkci krevních destiček; chronická trombocytopenie kvůli jejímu nedostatku.“ (PDF). Krev. 16 (1): 943–57. doi:10.1182 / krev.V16.1.943.943. PMID 14443744.
- ^ Upshaw JD (červen 1978). „Vrozený nedostatek faktoru v normální plazmě, který zvrací mikroangiopatickou hemolýzu a trombocytopenii“. N. Engl. J. Med. 298 (24): 1350–2. doi:10.1056 / NEJM197806152982407. PMID 651994.
- ^ A b Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, Sarode R, Shurin SB, Chandrasekaran V, Stabler SP, Sabio H, Bouhassira EE, Upshaw JD , Ginsburg D, Tsai HM a kol. (Říjen 2001). „Mutace u člena rodiny genů ADAMTS způsobují trombotickou trombocytopenickou purpuru“ (PDF). Příroda. 413 (6855): 488–494. Bibcode:2001 Natur.413..488L. doi:10.1038/35097008. hdl:2027.42/62592. PMID 11586351. S2CID 4380010.
- ^ Tsai HM (2009). „Trombotická trombocytopenická purpura, hemolyticko-uremický syndrom a související poruchy“. V publikaci Greer JP, Foerster J, Rodgers GM a kol. (eds.). Wintrobeova klinická hematologie (12. vydání). Philadelphia PA: Lippincott, Williams a Wilkins. str. 1314–25. ISBN 978-0781765077.
- ^ Kokame, K .; Matsumoto, M; Soejima, K; Yagi, H; Ishizashi, H; Funato, M; Tamai, H; Konno, M; Kamide, K; Kawano, Y; Miyata, T; Fujimura, Y (14. srpna 2002). „Mutace a běžné polymorfismy v genu ADAMTS13 odpovědné za aktivitu proteázy štěpící von Willebrandův faktor“. Proc. Natl. Acad. Sci. USA. 99 (18): 11902–7. Bibcode:2002PNAS ... 9911902K. doi:10.1073 / pnas.172277399. PMC 129366. PMID 12181489.
- ^ Menkes, John H .; Sarnat, Harvey B .; Maria, Bernard L. (2006). „Trombotická trombocytopenická purpura a hemolyticko-uremický syndrom“. Dětská neurologie (7. vydání). Philadelphia: Lippincott Williams & Wilkins. p. 525. ISBN 9780781751049.
- ^ van Mourik JA, Boertjes R, Huisveld IA a kol. (Červenec 1999). „propeptid von Willebrandova faktoru při vaskulárních poruchách: nástroj pro rozlišení mezi akutní a chronickou perturbací endoteliálních buněk“. Krev. 94 (1): 179–85. doi:10.1182 / krev.V94.1.179.413k18_179_185. PMID 10381511.
- ^ Iwata H, Kami M, Hori A, Hamaki T, Takeuchi K, Mutou Y (červen 2001). „Retrospektivní studie sekundární trombotické trombocytopenické purpury založená na pitvě“. Haematologica. 86 (6): 669–70. PMID 11418383.
- ^ A b C Noris M, Caprioli J, Bresin E a kol. (Říjen 2010). „Relativní role abnormalit genetického komplementu při sporadickém a familiárním aHUS a jejich dopad na klinický fenotyp“. Clin J Am Soc Nephrol. 5 (10): 1844–59. doi:10.2215 / CJN.02210310. PMC 2974386. PMID 20595690.
- ^ A b Neuhaus TJ, Calonder S, Leumann EP (červen 1997). „Heterogenita atypických hemolyticko-uremických syndromů“. Oblouk. Dis. Dítě. 76 (6): 518–21. doi:10.1136 / příd. 76.6.518. PMC 1717216. PMID 9245850.
- ^ A b C Dragon-Durey MA, Sethi SK, Bagga A a kol. (Prosinec 2010). „Klinické příznaky hemolyticko-uremického syndromu spojeného s autoprotilátkami proti faktoru H“. J. Am. Soc. Nephrol. 21 (12): 2180–7. doi:10.1681 / ASN.2010030315. PMC 3014031. PMID 21051740.
- ^ A b Caprioli J, Noris M, Brioschi S a kol. (Srpen 2006). „Genetika HUS: dopad mutací MCP, CFH a IF na klinický obraz, odpověď na léčbu a výsledek“. Krev. 108 (4): 1267–79. doi:10.1182 / krev-2005-10-007252. PMC 1895874. PMID 16621965.
- ^ A b Ariceta G, Besbas N, Johnson S a kol. (Duben 2009). „Pokyny pro vyšetřování a počáteční léčbu hemolyticko-uremického syndromu s negativním průjmem“. Pediatr. Nephrol. 24 (4): 687–96. doi:10.1007 / s00467-008-0964-1. PMID 18800230.
- ^ Al-Akash SI, Almond PS, Savell VH, Gharaybeh SI, Hogue C (duben 2011). „Eculizumab indukuje dlouhodobou remisi u rekurentního posttransplantačního HUS spojeného s mutací genu C3“. Pediatr. Nephrol. 26 (4): 613–9. doi:10.1007 / s00467-010-1708-6. PMID 21125405. S2CID 22334044.
- ^ Zuber J, Le Quintrec M, Sberro-Soussan R, Loirat C, Frémeaux-Bacchi V, Legendre C (leden 2011). "Nové poznatky o postrenálním transplantačním hemolyticko-uremickém syndromu". Nat Rev Nephrol. 7 (1): 23–35. doi:10.1038 / nrneph.2010.155. PMID 21102542. S2CID 2054556.
- ^ A b Tsai HM (leden 2010). "Patofyziologie trombotické trombocytopenické purpury". Int. J. Hematol. 91 (1): 1–19. doi:10.1007 / s12185-009-0476-1. PMC 3159000. PMID 20058209.
- ^ Bitzan M, Schaefer F, Reymond D (září 2010). "Léčba typického (enteropatického) hemolyticko-uremického syndromu". Semin. Tromb. Hemost. 36 (6): 594–610. doi:10.1055 / s-0030-1262881. PMID 20865636.
- ^ A b Michael, M; Elliott, EJ; Ridley, GF; Hodson, EM; Craig, JC (21. ledna 2009). „Intervence pro hemolyticko-uremický syndrom a trombotickou trombocytopenickou purpuru“. Cochrane Database of Systematic Reviews (1): CD003595. doi:10.1002 / 14651858.CD003595.pub2. hdl:10072/61440. PMC 7154575. PMID 19160220.
- ^ A b C d E Lim W, Veselý SK, George JN (5. března 2015). „Role rituximabu v léčbě pacientů se získanou trombotickou trombocytopenickou purpurou“. Krev. 125 (10): 1526–31. doi:10.1182 / krev-2014-10-559211. PMC 4351502. PMID 25573992.
- ^ A b Allford SL, Hunt BJ, Rose P, Machin SJ (únor 2003). „Pokyny k diagnostice a léčbě trombotických mikroangiopatických hemolytických anemií“. Br. J. Haematol. 120 (4): 556–73. doi:10.1046 / j.1365-2141.2003.04049.x. PMID 12588343. S2CID 38774829.
- ^ Peyvandi, Flora; Scullyová, Marie; Kremer Hovinga, Johanna A .; Cataland, Spero; Knöbl, Paul; Wu, Haifeng; Artoni, Andrea; Westwood, John-Paul; Mansouri Taleghani, Magnus (11.02.2016). „Caplacizumab pro získanou trombotickou trombocytopenickou purpuru“ (PDF). New England Journal of Medicine. 374 (6): 511–522. doi:10.1056 / NEJMoa1505533. ISSN 0028-4793. PMID 26863353.
- ^ Loirat, C; Girma, JP; Desconclois, C; Coppo, P; Veyradier, A (leden 2009). „Trombotická trombocytopenická purpura související se závažným nedostatkem ADAMTS13 u dětí“. Dětská nefrologie (Berlín, Německo). 24 (1): 19–29. doi:10.1007 / s00467-008-0863-5. PMID 18574602. S2CID 22209831.
- ^ Tsai, Han-Mou (únor 2006). „Současné pojmy v trombotické trombocytopenické purpuře“. Roční přehled medicíny. 57: 419–436. doi:10.1146 / annurev.med.57.061804.084505. PMC 2426955. PMID 16409158.
- ^ Terrell DR, Williams LA, Veselý SK, Lämmle B, Hovinga JA, George JN (červenec 2005). „Výskyt trombotického trombocytopenického purpura-hemolyticko-uremického syndromu: všichni pacienti, idiopatičtí pacienti a pacienti se závažným nedostatkem ADAMTS-13“. J. Thromb. Haemost. 3 (7): 1432–6. doi:10.1111 / j.1538-7836.2005.01436.x. PMID 15978100.
- ^ Terrell DR, Veselý SK, Kremer Hovinga JA, Lämmle B, George JN (listopad 2010). „Různé rozdíly mezi pohlavími a rasami mezi trombotickou trombocytopenickou purpurou a hemolyticko-uremickými syndromy“. Dopoledne. J. Hematol. 85 (11): 844–7. doi:10.1002 / ajh.21833. PMC 3420337. PMID 20799358.
- ^ X. Long Zheng; J. Evan Sadler (2008). „Patogeneze trombotických mikroangiopatií“. Výroční přehled patologie. 3: 249–277. doi:10.1146 / annurev.pathmechdis.3.121806.154311. PMC 2582586. PMID 18215115.
- ^ Moschcowitz E (1924). „Akutní febrilní pleiochromní anémie s hyalinní trombózou terminálních arteriol a kapilár: nepopsaná nemoc“. Proc NY Pathol Soc. 24: 21–4. Přetištěno Moschcowitz E (říjen 2003). „Akutní febrilní pleiochromní anémie s hyalinní trombózou terminálních arteriol a kapilár: nepopsaná nemoc. 1925“. Mt. Sinai J. Med. 70 (5): 352–5. PMID 14631522..
- ^ Amorosi EL, Ultmann JE (1966). „Trombocytopická purpura: zpráva o 16 případech a přehled literatury“. Medicína (Baltimore). 45 (2): 139–159. doi:10.1097/00005792-196603000-00003. S2CID 71943329.
- ^ A b Sadler, JE (2008). „Von Willerbrandův faktor, ADAMTS13 a trombotická trombocytopenická purpura“. Krev. 112 (1): 11–18. doi:10.1182 / krev-2008-02-078170. PMC 2435681. PMID 18574040.
- ^ Rock GA, Shumak KH, Buskard NA a kol. (Srpen 1991). „Srovnání výměny plazmy s plazmatickou infuzí při léčbě trombotické trombocytopenické purpury. Kanadská studijní skupina pro aferézu“. N. Engl. J. Med. 325 (6): 393–7. doi:10.1056 / NEJM199108083250604. PMID 2062330.
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |