Celková syntéza vitaminu B12 - Vitamin B12 total synthesis
tento článek může obsahovat nadměrné množství složitých detailů, které mohou zajímat pouze konkrétní publikum. (Červen 2020) (Zjistěte, jak a kdy odstranit tuto zprávu šablony) |
The celková syntéza komplexní biomolekuly vitamin B12 bylo dosaženo ve dvou různých přístupech spolupracujícími výzkumnými skupinami Robert Burns Woodward v Harvard[1][2][3][4][5] a Albert Eschenmoser v ETH[6][7][8][9][10][11][12] v roce 1972. Úspěch vyžadoval úsilí ne méně než 91 postdoktorandští vědci (Harvard: 77, ETH: 14)[13]:9-10[14]a 12 Ph.D. studenti (na ETH[12]:1420) z 19 různých národů po dobu téměř 12 let.[5](1:14:00-1:14:32,1:15:50-1:19:35)[14]:17-18 Syntetický projekt[15] vyvolala a zahrnovala zásadní změnu paradigma[16][17]:37[18]:1488 v oblasti přírodní produkt syntéza.[19][20][21]
Molekula

Vitamin B12, C.63H88Ošidit14Ó14P, je nejsložitější ze všech známých vitamíny. Jeho chemická struktura byla určena rentgenová krystalová strukturní analýza v roce 1956 výzkumnou skupinou Dorothy Hodgkin (Oxfordská univerzita ) ve spolupráci s Kenneth N. Trueblood v UCLA a John G. White v Univerzita Princeton.[22][23]Jádro molekuly je corrin struktura, dusíkatá tetradentátový ligand Systém.[poznámka 1] Tohle je biogeneticky související s porfyriny a chlorofyly, přesto se od nich v důležitých ohledech liší: uhlíkové kostře postrádá jeden ze čtyř mezokarbonů mezi pětičlennými kruhy, přičemž dva kruhy (A a D, obr. 1) jsou přímo spojeny jednoduchá vazba uhlík-uhlík. Corrin chromofor systém je tedy necyklický a rozšiřuje se pouze do tří poloh mezo a zahrnuje tři vinylogický amidin Jednotky. Seřadili na okraji makrocyklický prsten je osm methyl skupiny a čtyři propionální a tři octová kyselina boční řetězy. Devět atomů uhlíku na okraji corrinu je chirogenní centra. Tetradentát, jednosytný corrin ligand je ekvatoriálně koordinovaný s trojmocným kobalt ion, který nese dva další axiální ligandy.[poznámka 2]

Několik přirozených variant B12 existuje struktura, která se liší v těchto axiálních ligandech. V samotném vitaminu kobalt nese a kyano skupina na horní straně corrinovy roviny (kyanokobalamin ) a a nukleotid smyčka na druhé straně. Tato smyčka je připojena na svém druhém konci k periferní propionové amidové skupině na kruhu D a skládá se ze strukturních prvků odvozených od aminopropanol, fosfát, ribóza, a 5,6-dimethylbenzimidazol. Jeden z atomů dusíku v imidazol kruh je axiálně koordinován s kobaltem, nukleotidovou smyčkou, čímž vytváří devatenáctičlenný kruh. Všechny karboxylové skupiny postranního řetězce jsou amidy.
Kyselina kobyrová, jeden z přírodních derivátů vitaminu B.12,[24] postrádá nukleotidovou smyčku; v závislosti na povaze těchto dvou axiálních ligandů místo toho zobrazuje svoji funkci kyseliny propionové na kruhu D jako karboxylát (jak je znázorněno na obr. 1) nebo karboxylová kyselina (se dvěma kyanidovými ligandy na kobaltu).
Tyto dvě syntézy
Struktura vitamin B12 byla první nízkomolekulární hmotností přírodní produkt stanoveno spíše rentgenovou analýzou než chemickou degradací. Takže zatímco struktura tohoto nového typu komplexu biomolekula byla založena, její chemie zůstala v podstatě neznámá; zkoumání této chemie se stalo jedním z úkolů vitaminů chemická syntéza.[12]:1411[18]:1488-1489[25]:275 V 60. letech 20. století představovala syntéza takové výjimečně složité a jedinečné struktury hlavní výzvu na hranici výzkumu v oblasti syntézy organických přírodních produktů.[17]:27-28[1]:519-521


Již v roce 1960, výzkumná skupina biochemik Konrad Bernhauer v Stuttgart rekonstituoval vitamin B12 z jednoho z jeho přirozeně se vyskytujících derivátů, kyseliny cobyrové,[24] postupnou konstrukcí nukleotidové smyčky vitaminu.[poznámka 4] Tato práce činila a parciální syntéza vitaminu B12 z přírodního produktu obsahujícího všechny strukturní prvky vitaminu B12 kromě nukleotid smyčka. Proto byla jako cílová molekula pro celkovou syntézu vitaminu B vybrána kyselina cobyrová12.[6]:183-184[1]:521[8]:367-368
Spolupráce[3]:1456[17][28]:302-313 výzkumných skupin v Harvard a v ETH vyústil ve dvě syntézy kyseliny cobyrické, které byly současně provedeny v roce 1972,[29][30] jeden na Harvardu[3]a druhý na ETH.[10][11][12] „Konkurenční spolupráce“[17]:30[31]:626 o této velikosti, která zahrnuje 103 postgraduálních studentů a postdoktorandských výzkumných pracovníků, celkem tedy téměř 177 člověk-let,[13]:9-10 je zatím jedinečný v historii organická syntéza.[4](0:36:25-0:37:37) Tyto dvě syntézy jsou chemicky složitě propletené,[18]:1571 přesto se liší v zásadě způsobem centrálním makrocyklický je vytvořen systém ligandů corrin. Obě strategie jsou vzorovány po dvou modelových syntézách corrinů vyvinutých na ETH.[8][18]:1496,1499[32]:71-72 První, publikovaný v roce 1964,[26] dosáhl konstrukce chromoforu corrin kombinací A-D-složky s B-C-složkou via iminoester /enamin -C, C-kondenzace, konečného uzávěru corrinových kroužků je dosaženo mezi kroužky A a B.[33] Syntéza druhého modelu, publikovaná v roce 1969,[34] prozkoumal román fotochemické proces cykloisomerizace k vytvoření přímého spojení A / D-kruhu jako konečné uzavření corrinového kruhu mezi kruhy A a D.[35]
Přístup A / B k syntéze kyseliny cobyrické byl společně sledován a dokončen v roce 1972 na Harvardu. Spojilo to bicyklickou Harvardská A-D složka s ETH B-C složka a uzavřel makrocyklický corrinový kruh mezi kruhy A a B.[3]:145,176[4](0:36:25-0:37:37) A / D přístup k syntéze, provedený na ETH a dokončený současně s A / B přístupem také v roce 1972, postupně přidává kroužky D a A ke složce B-C přístupu A / B a dosáhne corrinova prstence uzávěr mezi kroužky A a D.[10][11][12] Cesty těchto dvou syntéz se setkaly ve společném korinoidním meziproduktu.[11]:519[36]:172 The poslední kroky z tohoto meziproduktu na kyselinu cobyrovou byly provedeny ve dvou laboratořích opět společně, přičemž každá skupina pracovala s materiálem připraveným vlastním přístupem.[17]:33[18]:1567
Synopse spolupráce Harvard / ETH
Počátky
Woodward a Eschenmoser se pustil do projektu chemické syntézy vitaminu B.12 nezávisle na sobě. Skupina ETH začala modelovou studií o tom, jak syntetizovat systém ligandů corrin v prosinci 1959.[18]:1501 V srpnu 1961[17]:29[13]:7 skupina Harvard začala útočit na hromadění B12 strukturu přímo zaměřením na nejsložitější část B12 molekula, „západní polovina“[1]:539 který obsahuje přímé spojení mezi kroužky A a D (složka A-D). Již v říjnu 1960[17]:29[13]:7[37]:67 skupina ETH zahájila syntézu prekurzoru vitaminu B v kruhu B12.
Na začátku,[38] pokrok na Harvardu byl rychlý, dokud projekt nepřerušil neočekávaný stereochemický průběh kroku formování centrálního prstence.[39][17]:29 Woodwardovo uznání stereochemické záhady, která vyšla najevo dráždivým chováním jednoho z jeho pečlivě naplánovaných syntetických kroků, se podle jeho vlastních spisů stalo[39] část vývoje, který vedl k pravidla orbitální symetrie.
Po roce 1965 skupina Harvard pokračovala v práci směrem k A-D-komponenta podle upraveného plánu pomocí (-) - kafr[40] jako zdroj prstenu D.[17]:29[18]:1556
Spojovací síly: přístup A / B k syntéze kyseliny cobyrické
Do roku 1964 skupina ETH dosáhla první corrin syntéza modelu,[26][25]:275 a také příprava prekurzoru kruhu B jako součást konstrukce B12 samotná molekula.[37][41] Protože nezávislý pokrok obou skupin směrem k jejich dlouhodobému cíli byl tak jasně doplňkový, rozhodli se Woodward a Eschenmoser v roce 1965[18]:1497[17]:30 spojit síly a od té doby pokračovat v projektu B.12 syntéza společně, plánování využití strategie konstrukce ligandu (prstenová vazba komponent) modelového systému ETH.[2]:283[18]:1555-1574
V roce 1966 se skupině ETH podařilo syntetizovat B-C složka („východní polovina“[1]:539) spojením jejich prekurzoru ring-B s prekurzorem ring-C.[18]:1557 Ten byl také připraven na Harvardu z (-) - kafru strategií koncipovanou a použitou dříve A. Pelterem a J. W. Cornforth v roce 1961.[poznámka 6] Na ETH zahrnovala syntéza B-C-komponenty implementaci C, C-kondenzační reakce přes sulfidová kontrakce. Ukázalo se, že tato nově vyvinutá metoda poskytuje obecné řešení problému konstrukce charakteristických strukturních prvků chrominoforu corrin, vinylogických amidinových systémů, které spojují čtyři periferní kruhy.[18]:1499
Na začátku roku 1967 uskutečnila skupina Harvard syntézu modelové A-D komponenty,[poznámka 7] s nediferencovaným řetězcem na straně f nesoucím methylesterovou funkci jako všechny ostatní boční řetězce.[18]:1557 Od té doby si obě skupiny systematicky vyměňovaly vzorky svých příslušných polovin corrinoidové cílové struktury.[17]:30-31[18]:1561[30]:17 Do roku 1970 společně spojili Harvardovu nediferencovanou A-D složku s ETH B-C složkou a vyrobili dikyano-kobalt (III) -5,15-bisnor-heptamethyl-kobyrinát 1 (obr. 4).[poznámka 2] Skupina ETH identifikovala tento zcela syntetický corrinoidový meziprodukt přímým porovnáním se vzorkem vyrobeným z přírodního vitaminu B.12.[2]:301-303[18]:1563
V této pokročilé modelové studii byly reakční podmínky pro náročné procesy Spojení C / D a cyklizace A / B pomocí metody sulfidové kontrakce. Ty pro C / D vazbu byly úspěšně prozkoumány v obou laboratořích, lepší podmínky byly ty, které byly nalezeny na Harvardu,[2]:290-292[18]:1562 zatímco metoda pro uzavření A / B-kroužku pomocí intramolekulární verze sulfidová kontrakce[44][34][45] byl vyvinut na ETH.[2]:297-299[46][18]:1562-1564 Později se na Harvardu ukázalo, že uzavření A / B-kroužku lze dosáhnout také thio-kondenzace iminoester / enamin.[2]:299-300[18]:1564
Na začátku roku 1971 dokončila skupina Harvard syntézu finální A-D složky,[poznámka 8] obsahující karboxylovou funkci na straně f na kruhu D odlišenou od všech karboxylových funkcí jako nitrilová skupina (jak je uvedeno v 2 v obr. 4; viz také obr. 3 ).[3]:153-157 A / D část B12 struktura zahrnuje ústavně a konfiguračně nejsložitější část molekuly vitaminu; jeho syntéza je považována za zbožnění Woodwardovského umění v celkové syntéze přírodních produktů.[11]:519[12]:1413[18]:1564[31]:626
Alternativní přístup k syntéze kyseliny cobyrické
Už v roce 1966[35]:1946 skupina ETH začala znovu v modelovém systému prozkoumávat alternativní strategii syntézy corrinů, při které by byl corrinový kruh uzavřen mezi kruhy A a D. Projekt byl inspirován představitelnou existencí dosud neznámé reoganizace vazby proces.[35]:1943-1946 To - pokud existuje - by umožnilo konstrukci kyseliny cobyrické z jediného výchozího materiálu.[6]:185[8]:392,394-395[31] Důležité je, že hypotetický proces, který je interpretován jako implikující dvě po sobě jdoucí přeskupení, byl formálně zahrnut do nových klasifikací reaktivity sigmatropních přesmyků a elektrocyklizací navržených Woodward a Hoffmann v kontextu jejich pravidla orbitální symetrie![8]:395-397,399[11]:521[47][18]:1571-1572
Do května 1968,[18]:1555 skupina ETH prokázala v modelové studii, že předpokládaný proces, fotochemická A / D-seco-korinát → korinátová cykloizomerace, ve skutečnosti existuje. Nejprve bylo zjištěno, že tento proces probíhá s komplexem Pd, ale vůbec ne s odpovídajícími komplexy Ni (II) - nebo kobalt (III) -A / D-seco-korinát.[34][48]:21-22 Rovněž probíhalo hladce v komplexech kovových iontů, jako je zinek a jiné fotochemicky inertní a volně vázané kovové ionty.[8]:400-404[12]:1414 Po uzavření kroužku je lze snadno nahradit kobaltem.[8]:404 Tyto objevy otevřely dveře tomu, co se nakonec stalo fotochemický A / D přístup syntézy kyseliny cobyrické.[7]:31[9]:72-74[35]:1948-1959

Počínaje podzimem 1969[49]:23 s B-C složka přístupu A / B a prekurzor ring-D připravený z enantiomer výchozího materiálu vedoucího k prekurzoru ring-B trvalo doktoranda Waltera Fuhrera[49] méně než jeden a půl roku[17]:32 převést fotochemický model syntézy corrinu na syntézu dikyano-kobaltu (III) -5,15-bisnor-a, b, d, e, g-pentamethyl-cobyrinátu-c-N, N-dimethylamid-f-nitril 2 (obr. 4 ), běžný korinoidní meziprodukt na cestě ke kyselině kobyrové. Na Harvardu, ten samý meziprodukt 2 bylo získáno přibližně ve stejné době spojením Harvard-D-diferencované Harvard-D-komponenty (dostupné na jaře 1971)[18]:1564 poznámka pod čarou 54a[3]:153-157) se složkou ETH B-C, použitím kondenzačních metod vyvinutých dříve s použitím nediferencované složky A-D.[1]:544-547[2]:285-300
Na jaře 1971 tedy[31]:634 dvě různé cesty ke společnému korininoidnímu meziproduktu 2 (obr. 4 ) na cestě ke kyselině kobyrové byla k dispozici, přičemž jeden vyžadoval 62 chemických kroků (Harvard / ETH A / B přístup ), dalších 42 (ETH A / D přístup ). V obou přístupech byly odvozeny čtyři periferní prstence enantiopure prekurzory se správným významem chirální čímž se obchází hlavní stereochemické problémy při tvorbě ligandového systému.[1]:520-521[7]:12-13[11]:521-522 Při konstrukci křižovatky A / D pomocí A / D-secocorrinu →corrin cykloisomerizace, tvorba dvou A / D-diastereomery muselo se očekávat. Použití kadmia (II) jako koordinačního kovového iontu vedlo k velmi vysoké diastereoselektivitě[49]:44-46 ve prospěch přirozeného A / D-trans-izomer.[12]:1414-1415
Jakmile byla corrinová struktura vytvořena kterýmkoli přístupem, tři C-H-chirogenní centra na okraji sousedícím s chromofor systém se ukázal být náchylný k epimerizace s výjimečnou lehkostí.[2]:286[9]:88[3]:158[4](1:53:33-1:54:08)[18]:1567 To vyžadovalo oddělení diastereomerů po většině chemických kroků v této pokročilé fázi syntéz. Mělo štěstí, že právě v té době byla technika vysokotlaká kapalinová chromatografie (HPLC) byl vyvinut v analytické chemii.[50] HPLC se stala nepostradatelným nástrojem v obou laboratořích;[30]:25[9]:88-89[3]:165[4](0:01:52-0:02:00,2:09:04-2:09:32) jeho použití v B.12 projekt propagovaný Jakobem Schreiberem na ETH,[51] byla první aplikací této techniky při syntéze přírodních produktů.[18]:1566-1567[36]:190[52]
Společné závěrečné kroky
The konečná konverze běžného korininoidního meziproduktu 2 (obr.6) ze dvou přístupů do cílové kyseliny kobylové vyžadovalo zavedení dvou chybějících methylové skupiny v mezo polohách chromoforu corrin mezi kruhy A / B a C / D, stejně jako konverze všech periferních karboxylových funkcí do jejich amidové formy, s výjimkou kritického karboxylu na řetězci na straně f kruhu D (viz obr. 6). Tyto kroky byly společně prozkoumány striktně paralelním způsobem v obou laboratořích, na Harvardově skupině s použitím materiálu vyrobeného přístupem A / B, skupina ETH připravená pomocí fotochemického přístupu A / D.[17]:33[18]:1567

První rozhodující identifikace a zcela syntetický středně pokročilí na cestě ke kyselině kobyrové byla provedena v únoru 1972 s krystalickým vzorkem zcela syntetického dikyanokobaltnatého (III) -hexamethyl-kobyrinátu-f-amidu 3 (obr[poznámka 2]), u kterých byla zjištěna totožnost ve všech datech s krystalickým reléovým vzorkem vyrobeným z vitaminu B.12 methanolýzou na kobester 4,[poznámka 9] následuje částečná amonolýza a separace výsledné směsi.[53]:44-45,126-143[3]:170[55]:46-47 V době, kdy Woodward oznámil „Totální syntézu vitaminu B12„na konferenci IUPAC v Novém Dillí v únoru 1972,[3]:177 zcela syntetický vzorek byl připraven na ETH fotochemickým A / D přístupem,[17]:35[56]:148[18]:1569-1570 zatímco první vzorek syntetické kyseliny cobyrové, identifikovaný s přírodní kyselinou cobyrickou, byl vyroben na Harvardu od B12- odvozený materiál f-amidového relé.[55]:46-47[3]:171-176 Úspěchem Woodward / Eschenmoser v té době tedy byly, přísně vzato, dvě formální totální syntézy kyseliny cobyricové a formální totální syntéza vitaminu.[55]:46-47[18]:1569-1570
V pozdějším průběhu roku 1972 dva krystalické epimery ze zcela syntetického dikyano-kobaltu (III) -hexamethyl-kobyrinátu-f-amide 3, stejně jako dva krystalické epimery zcela syntetického f-nitrilu, všechny připravené oběma syntetickými přístupy, byly přísně identifikován chromatograficky a spektroskopicky s odpovídajícím B12-odvozené látky.[18]:1570-1571[53]:181-197,206-221[5](0:21:13-0:46:32,0:51:45-0:52:49)[57] Na Harvardu se poté kyselina cobyrová vyráběla také ze zcela syntetického f-amidu 3 připraveno pomocí A / B přístupu.[55]:48-49 A konečně, v roce 1976 na Harvardu,[55] zcela syntetická kyselina cobyrová byla přeměněna na vitamin B.12 cestou průkopníkem Konrad Bernhauer.[poznámka 4]
Záznam publikace
Během téměř 12 let oběma skupinám trvalo, než dosáhly svého cíle, a to jak Woodward, tak Eschenmoser pravidelně přednášeli o fázi projektu spolupráce na přednáškách, některé z nich se objevily v tisku. Woodward diskutovali o přístupu A / B na přednáškách publikovaných v roce 1968,[1] a 1971,[2] který vyvrcholil oznámením „Celková syntéza vitaminu B12„v Dillí v únoru 1972[3]:177 publikováno v roce 1973.[3] Tato publikace a přednášky se stejným názvem přednesl Woodward v pozdější části roku 1972[4][5] jsou omezeny na A / B přístup syntézy a nediskutují o ETH A / D přístupu.
Eschenmoser diskutoval o příspěvcích ETH k přístupu A / B v roce 1968 ve 22 Robert A. Welch Foundation konference v Houstonu,[7] stejně jako v jeho 1969 Přednáška RSC Centenary „Roads to Corrins“, publikováno v roce 1970.[8] Představil fotochemický A / D přístup ETH k B12 syntéza na 23 IUPAC Kongres v Bostonu v roce 1971.[9] Curyšská skupina oznámila dokončení syntézy kyseliny cobyrické fotochemickým A / D přístupem ve dvou přednáškách, které přednesli doktorandi Maag a Fuhrer na zasedání Švýcarské chemické společnosti v dubnu 1972,[10] Eschenmoser přednesl přednášku „Celková syntéza vitaminu B12: Photochemical Route “poprvé jako přednáška Wilsona Bakera na univerzitě v Bristolu v Bristolu ve Velké Británii 8. května 1972.[poznámka 10]


Jako společná úplná publikace syntéz skupin Harvard a ETH (oznámeno v[10] a očekává se v[11]) se neobjevil do roku 1977,[poznámka 12] článek popisující finální verzi fotochemického A / D přístupu provedeného již v roce 1972[10][49][53][61] byl publikován 1977 v Science.[12][56]:148 Tento článek je rozšířeným anglickým překladem jednoho, který se již objevil v roce 1974 v Naturwissenschaften,[11] na základě přednášky Eschenmosera 21. ledna 1974 na zasedání Zürcher Naturforschende Gesellschaft. O čtyři desetiletí později, v roce 2015, tentýž autor konečně publikoval sérii šesti úplných článků popisujících práci skupiny ETH corrin syntéza.[62][18][63][64][33][35] Část I série obsahuje kapitolu nazvanou „Poslední fáze spolupráce Harvard / ETH na syntéze vitaminu B12",[18]:1555-1574 ve kterém jsou příspěvky skupiny ETH ke společné práci na syntéze vitaminu B.12 mezi lety 1965 a 1972.
Celá ETH práce je podrobně dokumentována ve veřejně přístupném Ph.D. práce,[37][41][58][44][59][54][60][42][46][49][53][61] téměř 1 900 stránek, vše v němčině.[65] Do těchto tezí jsou většinou integrovány příspěvky 14 postdoktorských výzkumníků ETH, kteří se podílejí na syntéze kyseliny cobyrické.[12]:1420[62]:1480[13]:12,38 Podrobná experimentální práce na Harvard bylo dokumentováno ve zprávách 77 zúčastněných postdoktorských výzkumníků s celkovým objemem více než 3 000 stránek.[13]:9,38[poznámka 11]
Reprezentativní recenze dvou přístupů k chemické syntéze vitaminu B.12 byly podrobně publikovány A. H. Jacksonem a K. M. Smithem,[43] T. Goto,[66] R. V. Stevens,[36] K. C. Nicolaou & E. G. Sorensen,[15][19] shrnul J. Mulzer & D. Riether,[67] a G. W. Craig,[14][31] kromě mnoha dalších publikací, kde jsou tyto epochální syntézy diskutovány.[poznámka 13]
Harvardský / ETH přístup k syntéze kyseliny cobyrické: cesta ke společnému korininoidnímu meziproduktu prostřednictvím uzávěru A / B-corrinového kruhu
Při přístupu A / B ke kyselině cobyronové byla Harvardova A-D složka spojena s ETH B-C složka mezi kruhy D a C a poté uzavřeny do korinů mezi kruhy A a B. Oba tyto kritické kroky byly provedeny C, C vazba přes sulfidovou kontrakci, nový typ reakce vyvinutý při syntéze B-C-komponenty na ETH. Složka A-D byla syntetizována na Harvardu z prekurzoru kruhu A (připraveného z achirál výchozí materiály) a prekurzor ring-D připravený z (-) - kafr. Ke zkoumání podmínek vazby byla použita modelová složka A-D; tato složka se lišila od složky A-D použité v konečné syntéze tím, že jako funkční skupina v řetězci f-postranního řetězce methylester skupina (jako všechny ostatní postranní řetězce) místo a nitril skupina.
| Harvardská syntéza A-D-komponent pro A / B přístup |
|---|
Syntéza prekurzoru kruhu A Obrázek 8: Harvardská syntéza složek A-D: kruh A Výchozím bodem pro syntézu prekurzoru kruhu-A byl methoxydimethyl-indol H-1 syntetizoval kondenzace z Schiffova základna z m-anisidin a acetoin. Reakce s Grignardovo činidlo z propargyl jodid dal racemický propargyl indolenin závod-H-2; uzavření kroužku do aminoketon závod-H-3 byl způsoben BF3 a HgO v MeOH přes meziprodukt závod-H-2a (elektrofilní přidání) se dvěma methylovými skupinami vynucenými do a cis-vztah kinetických i termodynamických důvodů.[1]:521-522  Obrázek 9: Harvardská syntéza složek A-D: rozlišení kruhu A. Rozlišení racemického aminoketonu do těchto dvou enantiomery. Reakce závod-H-3 s (-) - ethylem isokyanát povolená izolace do krystalizace jednoho ze dvou diastereomerní vytvořené deriváty močoviny (druhá nekrystalizuje). Léčba racemického ketonu závod-H-3 (nebo z matečné louhy z předchozí krystalizace) s (+) - ethylisokyanátem poskytl enantiomer prvního močovina derivát. Pyrolytický rozklad každého z těchto derivátů močoviny vedl k enantiopure aminoketony, požadované (+) - H-3, a (-) - H-3.[1]:524-525 „Nepřirozený“ (-) - enantiomer (-) - H-3 byl použit k určení absolutního konfigurace; v různých pozdějších krocích, (-) - H-3 a enantio-meziprodukty z něj odvozené byly použity jako modelové sloučeniny v průzkumných experimentech.[36]:173 Woodward napsal ohledně nepřirozeného enantiomeru „naše zkušenosti byly takové, že se jedná o jediný druh modelové studie, kterou považujeme za zcela spolehlivou“.[1]:529  Obrázek 10: Harvardova syntéza komponent A-D: Prsten A Určení konfigurace Stanovení absolutní konfigurace prekurzoru ring-A (+) - H-3. Pro toto stanovení je levotočivý („nepřirozený“) enantiomer aminoketonu (-) - H-3 byl použit k záchraně vzácného materiálu: Acylace aminoskupiny (-) - H-3 s chloracetylchlorid, po kterém následuje ošetření produktu H-3a s draslík t-butoxid v t-butanol, poskytl tetracyklický ketolaktam H-3b. Jeho ketokarbonyl byl převeden na methylenovou skupinu odsíření z dithioketal z H-3b s Raney nikl dát laktam H-3c. Zničení aromatického kruhu ozonolýza zahrnující spontánní ztrátu karboxylové funkce dekarboxylace, vedlo k bicyklické laktam-karboxylové kyselině H-3d. Tento materiál byl identifikován u produktu H-3h odvozený od (+) - kafr, které mají stejnou ústavu a absolutní konfiguraci, jak je uvedeno ve vzorci H-3d.[1]:525-526 Materiál pro tuto identifikaci H-3d byl syntetizován z (+) - kafru následujícím způsobem: cis-isoketopinová kyselina H-3e, získané z (+) - kafru zavedenou cestou popsanou v literatuře,[68] byl převeden prostřednictvím korespondenta chlorid, azid, a isokyanát na methyl-urethan H-3f. Při léčbě draslíkem t-butoxid dovnitř t-butanol a následně KOH, H-3f byl převeden na H-3h, jasně prostřednictvím meziproduktu H-3g. Totožnost dvou vzorků H-3d a H-3h získané dvěma popsanými cestami stanovilo absolutní konfiguraci (+) - H-3, enantiomer prekurzoru kruhu A.[1]:525-526 Syntéza prekurzoru ring-D z (-) - kafru  Obrázek 11: Harvardská syntéza složek A-D: kruh D z (-) - kafru (-) - Kafr byl nitrosovaný v a-poloze karbonylové skupiny dát oxim H-4, Beckmann štěpení poskytl přes odpovídající nitril amid H-5. Hofmannova degradace prostřednictvím zprostředkujícího aminu a jeho uzavření kruhu vedlo k laktamu H-6. Přeměna jeho N-nitroso derivát H-7 dal diazo sloučenina H-8. Tepelný rozklad H-8 indukovaný methyl migrace dát cyklopenten H-9. Snížení na H-10 (LiAlH4 ), oxidace (kyselina chromová ) na aldehyd H-11, Wittigova reakce (karbomethoxymethylenetrifenylfosforan ) až H-12 a hydrolýza esterové skupiny nakonec poskytla trans-karboxylová kyselina H-13.[1]:527-528[poznámka 14] Spojení prekurzorů ring-A a ring-D s "pentacykenonem"  Obrázek 12: Harvardova syntéza složek A-D: vazba kruhů A a D na „pentacylenon“ N-acylace tricyklického aminoketonu (+) - H-3 s chloridem H-14 karboxylové kyseliny H-13 dal amid H-15, který při léčbě draslíkem t-butoxid dovnitř t-butanol stereoselektivně produkoval pentacyklický keto-laktam H-16 přes intramolekulární Michaelova reakce který směruje indikované atomy vodíku navzájem trans. V očekávání Snížení břízy z aromatický kruh, ochranné skupiny pro ty dva karbonylové funkce z H-16 byly požadovány, jeden pro ketonovou karbonylovou skupinu jako ketal H-17a druhý pro laktam karbonyl jako vysoce citlivý enol ether H-20. Dopis ochrana bylo dosaženo léčbou H-17 s Meerweinová sůl (triethyloxonium tetrafluoroborát) dát iminová sůl H-18, následuje konverze na orthoamid H-19 (NaOMe / MeOH) a nakonec vyloučením jedné molekuly methanolu zahřátím v toluenu. Snížení břízy H-20 (lithium v kapalině amoniak, t-butanol, THF ) poskytl tetraen H-21. Zpracování kyselinou za pečlivě kontrolovaných podmínek vedlo nejprve k meziproduktu dione s dvojnou vazbou v poloze β, γ, která se přesunula do konjugované pozice v dione H-22dabovaný pentacykenon.[1]:528-531[14]:5 Od „pentacykenonu“ po „corrnorsteron“  Obrázek 13: Harvardská syntéza složek A-D: od „pentacylenonu“ po „corrnorsteronu“ Ethylenketal chránící skupina v pentacykenonu H-22 byl převeden na ketonovou skupinu H-23 kyselinou katalyzovaný hydrolýza.[1]:531 The dioxim primárně tvořen reakcí diketonu H-23 s hydroxylamoniumchlorid byl regioselektivně hydrolyzovaný (kyselina dusitá / kyselina octová) na požadovaný mono-oxim H-24. Toto je oxim stericky více bráněno ketonová skupina, jejíž atom dusíku je určen k tomu, aby se stal dusíkem kruhu cílové molekuly D. Rozhodující pro tento účel je konfigurace na monooximové dvojné vazbě zaujímá hydroxylová skupina stericky méně bráněnou pozici.[1]:532 Dvojné vazby C, C cyklopentenového a cyklohexenonového kruhu H-24 poté byly štěpeny ozonolýza (ozon při 80 ° C v MeOH, kyselina jodistá ) a vytvořená karboxylová skupina se esterifikuje CH2N2 ) na diketon H-25. An intramolekulární kondenzace aldolu 1,5-dikarbonylové jednotky v MeOH za použití pyrrolidin acetát jako základ, následovaný tosylace oximové hydroxylové skupiny poskytl derivát cyklohexenonu H-26. Druhá ozonolýza za mokra methylacetát, následovalo ošetření kyselinou jodistou a CH2N2 dal H-27. Beckmann přesmyk (MeOH, polystyrensulfonát sodný, 2 hodiny, 170 ° C) vznikl regioselektivně[1]:532 laktam H-27a (neizolovaný), který dále reagoval v amin-karbonylové kondenzaci → aldolová kondenzační kaskáda na tetracyklu H-28,[1]:533-534 volala α-corrnorsteron, implikovat to jako "základní kámen"[1]:534 při syntéze požadované A-D-složky.[1]:531-537 Tato sloučenina vyžadovala silně alkalické podmínky, aby se otevřela laktam prsten, ale bylo zjištěno, že nezletilý izomer, také izolované z reakční směsi, β-corrnorsteron H-29, prochází tímto laktamovým kruhem v zásaditém stavu velmi snadno.[1]:536 Strukturálně se oba izomery liší pouze v orientaci postranního řetězce kyseliny propionové na kruhu A: β-izomer má stabilnější trans-orientaci tohoto řetězce vzhledem k sousednímu řetězci kyseliny octové vytvořenému po otevření laktamového kruhu. Vyvážení α-corrnorsteronu H-28 zahřátím na silnou bázi, následovaným okyselením a zpracováním diazomethan, vedlo k izolaci čistého β-corrnorsteronu H-29 v 90% výtěžku.[1]:537 Správná absolutní konfigurace šesti contigous asymetrická centra v β-corrnorsteronu byla potvrzena pomocí rentgenová analýza krystalové struktury brom-p-corrnorsteronu[69][1]:529 s „nepřirozenou“ konfigurací.[1]:538[14]:8[4](0:49:20-0:50:42) Syntéza A-D-složky nesoucí funkci kyseliny propionové na kruhu D jako methoxykarbonylové skupiny (model A-D-složka)  Obrázek 14: Harvardova syntéza A-D-komponent: f-nediferencovaný model A-D-komponenty Léčba β-corrnorsteronu H-29 s methanolovou HC1 odštěpí laktamový kruh a získá se enol ether derivát s názvem hesperimin[poznámka 15] H-30u. Ozonolýza na aldehyd H-32u, redukce aldehydové skupiny s NaBH4 v MeOH do primární alkohol H-33u a nakonec přeměna hydroxyskupiny na odpovídající mesylát dal bromid H-34u. To představuje modelovou složku A-D, složku s funkcí nediferencované kyseliny propionové na kruhu D (tj. Nesoucí methylesterovou skupinu jako všechny ostatní postranní řetězce).[1]:539-540 Syntéza složky A-D nesoucí funkci kyseliny propionové na kruhu D jako nitrilové skupiny  Obrázek 15: Harvardská syntéza A-D-komponent: F-diferencovaná A-D-komponenta Konverze β-corrnorsteronu H-29 na správnou A-D složku H-34[1]:538-539 obsahující karboxylovou funkci postranního řetězce kyseliny propionové v kruhu D jako a nitril skupina, diferencovaná od všech ostatních methoxykarbonylových skupin, zahrnovala následující kroky: ošetření H-29 s methanolovým roztokem thiofenol a HC1 poskytly fenylthioenoletherový derivát H-30, který při ozonolýze při nízké teplotě poskytl odpovídající thioester -aldehyd H-31 a když následuje zpracování kapalným amoniakem, amid H-32. Redukce aldehydové skupiny pomocí NaBH4 na H-33, mesylace primární hydroxylové skupiny anhydrid kyseliny methansulfonové za podmínek, které také převádějí primární amidovou skupinu na požadovanou nitrilovou skupinu a nakonec nahrazení methansulfonyloxyskupiny bromidem produkovanou A-D složkou H-34 s funkcí kyseliny propionové na kruhu D jako nitril, odlišenou od všech ostatních takových postranních řetězců.[1]:539-540[4](1:01:56-1:19:47) |
| Spojení harvardských A-D komponent s ETH B-C komponentami |
|---|
Stavba corrin chromofor s jeho třemi vinylogický amidin jednotky tvoří - kromě přímého spojení jednoduchou vazbou mezi kruhy A a D - ústřední výzvu pro jakýkoli pokus o syntézu vitaminu B12. Úplně první přístup k úplné syntéze vitaminu B.12 zahájil Cornforth[43]:261-268 byl přerušen, když byl konfrontován s úkolem spojit syntetizované kruhové prekurzory.[18]:1493,1496 Spojení harvardských A-D komponent s ETH B-C komponentou vyžadovalo rozsáhlou průzkumnou práci, a to navzdory znalostem získaným v ETH modelových syntézách méně složitých (tj. Méně periferně substituovaných) korinů. To, co lze nazvat epickým střetnutím s formálním vytvořením pouze dvou vazeb C, C, trvalo od začátku roku 1967[18]:1557 do června 1970.[2] Na ETH i na Harvardu byly provedeny rozsáhlé modelové studie spojování zjednodušených enaminoid analogy A-D-složky s (kruh C) imino- a thioiminoesterový derivát plnohodnotné BC složky důsledně ukazoval, že spojení Harvardské a ETH složky lze jen těžko dosáhnout metodou, která byla tak úspěšná při syntéze jednodušších corrinů, konkrétně pomocí kondenzace intermolekulárního enamino-imino (nebo thio-imino) esteru[7][8][18]:1561[60]:41-58[1]:544[4](1:25:02-1:26:26) Výsledek těchto modelových studií určil konečný typ struktury harvardské A-D složky: struktura schopná působit jako součást C / D vazby sulfidová kontrakce prostřednictvím alkylační vazby,[8]:384-386[45] tj. bromid H-34u.[7]:18-22[60]:47,51-52 Tuto metodu již skupina ETH implementovala při syntéze B-C složka.[31]:16-19[35]:1927-1941[18]:1537-1540 Rozsáhlé hledání optimálních podmínek, nejprve pro C / D vazbu A-D složky s ETH B-C složkou E-19, poté byly v obou laboratořích sledovány podmínky následného intramolekulárního uzavření A / B-corrinového kruhu pomocí f-nediferencovaného modelu A-D složky[poznámka 7] H-34u[1]:540 jako model.[2]:287-300[18]:1561-1564 Jako výsledek práce od Yoshito Kishi na Harvardu,[2]:290[18]:1562[14]:11-12 a Peter Schneider na ETH,[46]:12,22-29[18]:1563-1564 optimální podmínky pro C / D-vazbu byly nakonec nalezeny na Harvardu, zatímco první a nejspolehlivější metoda pro uzavření korinového kruhu mezi kruhy A a B byla vyvinuta na ETH.[18]:1562 Postupy spojování C / D a uzávěru A / B-corrinového kruhu vyvinuté v této modelové řadě byly později aplikovány na odpovídající kroky v f-diferencovaná řada jako součást syntézy kyseliny kobyrové. Syntéza dikyano-kobaltu (III) -5,15-bisnor-a, b, c, d, e, f, g-heptamethyl-kobyrinátu z nediferencovaného modelu A-D složky kruhu-D D / C spojka.[7]:22-23[2]:287-292[46]:12,22-28[18]:1561-1562  Obrázek 16: Přístup Harvard / ETH A / B ke kyselině kobyrové: D / C vazba A-D složky Harvardského modelu s ETH B-C složkou Klíčovým problémem v tomto kroku byla labilita primárního spojovacího produktu, thioether HE-35u, izomerizující na jiné thioethery zpočátku nepodléhající sulfidové kontrakci v reprodukovatelném postupu s přijatelnými výtěžky.[2]:287-290[4](1:26:59-1:32:00) Vyvolávána draslíkem t-butoxid v THF /t-butanol za přísně kontrolovaných podmínek s přísným vyloučením vzduchu a vlhkosti, model A-D-komponenta H-34u plynule reagoval s B-C složkou E-19[46]:53-58 za vzniku spojovacího produktu přemostěného sírou HE-35u, pojmenovaný „thioether typu I“, v podstatě v kvantitativním výtěžku.[2]:287-288 Tento produkt však lze izolovat pouze za velmi pečlivě kontrolovaných podmínek, protože extrémně snadno ekvilibruje (např. Chromatografií nebo stopami kyseliny trifluoroctové v roztoku methylenchloridu) na stabilnější izomerní thioether HE-36u (thioether typu II), který na rozdíl od thioetheru typu I obsahuje π-systém konjugativně stabilizovaného vinylogického amidinu.[2]:289 V závislosti na podmínkách ještě další izomer HE-37u (thiother typu III).[2]:290 Počínaje takovými směsmi produktů kondenzace, při ETH různé podmínky (např. Komplex methyl-rtuť, BF3, trifenylfosfin[46]:58-65[2]:291) bylo zjištěno, že indukují (prostřednictvím HE-38u) krok kontrakce na HE-39u ve středních výnosech.[18]:1562[2]:287-292 S výběrem rozpouštědla, který byl shledán zásadním,[4](1:34:52-1:35:12) the optimal procedure at Harvard was heating thiother type II HE-36u v sulfolan in the presence of 5.3 equivalents kyselina trifluoroctová and 4.5 equivalents of tris-(β-cyanoethyl)-phosphine at 60 °C for 20 hours, producing HE-39u in up to 85% yield.[2]:292[46]:65-72 Later it was discovered that nitromethan could also be used as solvent.[4](1:34:52-1:35:13)[46]:28 A/B-ring closure.[2]:293-300[46]:12,29-39[18]:1562-1564 The problem of corrin-ring closure between rings A and B was solved in two different ways, one developed at ETH, the other pursued at Harvard.[30]:19 Both methods correspond to procedures developed before in the synthesis of metal complexes[70] as well as free ligands[71] of simpler corrins.[7]:25-28[8]:387-389[18]:1563 In the explorations of ring-closure procedures for the much more highly substituted A/B-seco-corrinoid intermediate HE-39u, the ETH group focused on the intramolecular version of the oxidative sulfide contraction method, eventually leading to the dicyano-cobalt(III)-complex HE48u.[46]:29-39[2]:297-299 This first totally synthetic corrinoid intermediate was identified with a corresponding sample derived from vitamin B12.[18]:1563 At Harvard, it was shown that the closure to the corrin macrocycle could also be realized by the method of thioiminoester/enamine condensation.[2]:299-300 All reactions described here had to be executed on a very small scale, with "... the utmost rigour in the exclusion of oxygen from the reaction mixtures"[2]:296, and most of them also under strict exclusion of moisture and light, demanding very high standards of experimental expertise.[2]:304 The major obstacle in achieving an A/B-corrin-ring closure was the exposure of the highly unstable ring B exocyklický methylidene double bond, which tends to isomerize into a more stable, unreactive endocyclic position with great ease.[46]:86,97-98[2]:293-294[3]:161[18]:1562  Figure 17: Harvard/ETH A/B approach to cobyric acid: A/B-ring closure to the f-undifferentiated model 5,15-bisnorcobyrinat The problem was solved at ETH[18]:1562-1563[46]:29-39,126-135 by finding that treatment of the thiolactone-thiolactam intermediate HE-40u (obtained from HE-39u by reacting with P2S5[46]:73-83) s dimethylamine in dry MeOH (room temperature, exclusion of air and light) smoothly opens the thiolakton ring at ring B, forming by elimination of H2S the exocyclic methylidene double bond as well as a dimethylamino-amide group in the acetic acid side chain.[46]:32-34,96-99 These conditions are mild enough to prevent double bond tautomerization to the thermodynamically more stable isomeric position in the ring. Immediate conversion with a Zn-perchlorate-hexa(dimethylformamide) complex in methanol to zinc complex HE-41u, followed by oxidative coupling (0,05 mM solution of Já2 /KI in MeOH, 3 h) afforded HE-42u.[46]:100-105 Sulfide contraction (triphenylphosphine, trifluoroacetic acid, 85 °C, exclusion of air and light) followed by re-complexation with Zn(ClO4)2 (KCl, MeOH, diisopropylamine ) led to the chloro-zinc complex HE-43u.[46]:105-116 The free corrinium salt formed when HE-43u was treated with trifluoroacetic acid in acetonitril was re-complexed with anhydrous CoCl2 in THF to the dicyano-cobalt(III)-complex HE-44u.[46]:117-125[2]:295 Conversion of the dimethylamino-amide group in the acetic acid side chain of ring B into the corresponding methylester group (Ó-methylation by trimethyloxonium tetrafluoroborate, followed by decomposition of the iminium salt with aqueous NaHCO3) afforded totally synthetic 5,15-bisnor-heptamethyl cobyrinate HE-48u.[46]:11,117-125 A crystalline sample of HE-48u was identified via UV/VIS, IR, a OBJEDNÁVKA spectra with a corresponding crystalline sample derived from vitamin B12[46]:42,135-141[53]:14,64-71,78-90[2]:287,301-303[3]:146-150[72] Later at Harvard,[2]:299-300 the A/B-corrin-ring closure was also achieved by converting the thiolactone-thiolactame intermediate HE-40u to thiolactone-thioiminoester HE-45u podle S-methylation of the thiolactam sulfur (MeHgOi-Pr, then trimethyloxonium tetrafluoroborate). Produkt HE-45u was subjected to treatment with dimethylamine (as in the ETH variant), forming the highly labile methylidene derivative HE-46u, which then was converted with anhydrous CoCl2 in THF to dicyano-cobalt(III) complex HE-47u, the substrate ready to undergo the (A⇒B)-ring closure by a thioiminoester/enamine condensation. A careful search at Harvard for reaction conditions led to a procedure (KO-t-Bu, 120 °C, two weeks) that gave corrin Co complex HE-44u, identical with and in overall yields comparable with HE-44u obtained by the ETH variant of the sulfide contraction procedure.[2]:300 Since in corrin model syntheses such a C,C-condensation required induction by a strong base, its application in a substrate containing seven methylester groups was not without problems;[18]:1562 in a, milder reactions conditions were applied.[3]:162 Synthesis of dicyano-cobalt(III)-5,15-bisnor-a,b,d,e,g-pentamethyl-cobyrinate-c-N, N-dimethylamide-f-nitrile (the common corrinoid intermediate) from the ring-D-differentiated A-D-component  Figure 18: Harvard/ETH A/B approach to cobyric acid: coupling of the Harvard f-differentiated A-D-component with the ETH B-C-component to the common corrinoid intermediate The A-D-component H-34[poznámka 8] with its propionic acid function at ring D differentiated from all the other carboxyl functions as nitrile group had become available at Harvard in spring 1971.[49]:23 As a result of the comprehensive exploratory work that had been done with the model A-D-component at Harvard and ETH,[2]:288-292[46]:22-28[18]:1561-1562 joining the proper A-D-component H-34 with the B-C-component E-19 by three operations H-34 + E-19 →→ HE-36 → HE-39.[3]:158-159[4](1:19:48-1:36:15) Closing the corrin ring was achieved in the sequence HE-39 (Str2S5, xylen, γ-picoline )→ HE-40[4](1:36:45-1:37:49) → HE-41[4](1:37:51-1:42:33) → HE-42[4](1:42:35-1:44:34) → HE-43 (overall yield "about 60 %"[4](1:44:35-1:46:32)), and finally to cobalt complex HE-44.[4](1:46:34-1:52:51)[3]:160-166 Reactions in this sequence were based on the procedures developed in the undifferentiated model series.[2]:293-300[46]:29-39[18]:1562-1564 Two methods were available for the A/B-ring closure: oxidative sulfide contraction within a zinc complex, followed by exchange of zinc by cobalt (ETH[3]:162-165), or the Harvard alkylative variant of a sulfide contraction,[3]:160-162 thio-iminoester /enamin condensation of the cobalt complex (improved reaction conditions: diazabicyclononanone in DMF, 60 °C, several hours[3]:162). Woodward preferred the former one:[3]:165 "...the oxidative method is somewhat superior, in that it is relatively easier to reproduce, .... ".[4](1:52:37-1:53:06) The corrin complex dicyano-cobalt(III)-5,15-bisnor-pentamethyl-cobyrinate-c-N, N-dimethylamide-f-nitrile HE-44 took up the role of the common corrinoid intermediate in the two approaches to cobyric acid synthesis: HE-44 ≡ E-37. Due to the high configurational lability of C-H chirogenic centers C-3, C-8 and C-13[4](1:21:49-1:23:42,1:35:43-1:36:14,1:51:51-1:52:30) na ligand periphery in basic or acidic milieu, separation by HPLC was indispensable for isolation, purification and characterization of pure diastereomers of this and the following corrinoid intermediates.[3]:165-166[9]:88-89[4](1:53:07-2:01:24) |
| Preparation of ring-C precursor from (+)-camphor by the Harvard group |
|---|
 Figure 19: Harvard preparation of the ring-C precursor from (+)-camphor Starting material for the synthesis of a ring-C precursor was (+)-camphorquinone H-35[poznámka 16] which was converted to the acetoxy-trimethylcyclohexene-carboxylic acid H-36 podle BF3 v anhydrid kyseliny octové, a reaction pioneered by Manasse & Samuel in 1902,[73], already successfully applied in a previous synthesis of the ring-C precursor by Pelter and Cornforth.[poznámka 6] Conversion of H-36 to amide H-37 was followed by its ozonolýza na peroxid H-38 which was reduced to the keto-sukcinimid H-46 by zinc and MeOH. Treatment with methanolic HCl gave lactam H-40, followed by thermal odstranění of methanol to the ring-C precursor H-41[1]:540-542[46]:49-50[14]:4-5,15 This was found to be identical with the ring-C precursor E-13 prepared by a different route[poznámka 5] at ETH.[59]:32[42]:30,33-34,81 |
The ETH approach to the synthesis of cobyric acid: the path to the common corrinoid intermediate via A/D-corrin-ring closure
In the A/D approach to the synthesis of cobyric acid, the four ring precursors (ring-C precursor only formally so[12]:ref. 22) derive from the two enantiomers of one common chirální starting material. All three vinylogous amidine bridges that connect the four peripheral rings were constructed by the sulfide contraction method, with the B-C-component – already prepared for the A/B-approach – serving as an intermediate.[12][11] The photochemical A/D-secocorrin→corrin cycloisomerization, by which the corrin ring was closed between rings A and D, is a novel process, targeted and found to exist in a model study (srov. obr. 2 ).[34][35]:1943-1948
| Synthesis of the ETH B-C-component (part of the A/B as well as A/D approach) |
|---|
Syntheses of the ring-B precursor Two syntheses of ring-B precursor (+)-E-5 were realized; the one starting from 2-butanone was used further.[6]:188 Two pathways for the conversion of the ring-B precursor into the ring-C precursor (+)-E-5 → (−)-E-13 ≡ H-41 were developed, one at ETH,[42]:15-39[1]:544, and one at Harvard.[6]:193[poznámka 17] These conversions turned out to be inadequate for producing large amounts of ring-C-precursor.[44]:38[18]:1561 However, the pathway developed at ETH served the purpose of determining the absolute configuration of the ring-B precursor.[6]:193[59]:32 Bulk amounts of ring-C precursor to be used for the production of the B-C-component at ETH[42]:40[6]:193[31]:631 were prepared at Harvard z (+)-camphor by a route originally developed by Pelter and Cornforth.[poznámka 6]  Figure 20: ETH synthesis of the B-C-component: synthesis of the two enantiomers of the ring-B precursor Ring-B precursor from 2-butanone and glyoxylic acid. Aldolová kondenzace mezi 2-butanon a kyselina glyoxylová by treatment with concentrated kyselina fosforečná ) gave stereoselectively (trans)-3-methyl-4-oxo-2-pentenoic acid E-1.[37]:11-20,45-45 Diels-Alder reaction of E-1 s butadien in benzene in the presence of SnCl4 afforded the racemát z chirální Diels-Alder adduct E-2 který byl vyřešen into the enantiomers by sequential salt formation with both (−)- and (+)-1-phenylethylamine.[41]:22,59-62 The chirogenic centers of the (+)-enantiomer (+)-E-2 possessed the absolute konfigurace z ring B in vitamin B12.[58]:35[6]:191 Oxidation of this (+)-enantiomer with kyselina chromová in acetone in the presence of kyselina sírová afforded the dilactone (+)-E-3 of the intermediary tricarboxylic acid E-3a.[41]:35,72-73 Thermodynamic control of dilactone formation leads to the cis-configuration of the ring junction.[41]:32-34 Elongation of the acetic acid side chain of (+)-E-3 podle Arndt-Eistert reaction (via the corresponding chlorid kyseliny and diazoketone) gave dilactone (+)-E-4.[59]:15-16,65-67 Léčba (+)-E-4 s NH3 in MeOH at room temperature formed a dual mixture of isomeric laktam -laktony in a ratio of 2:1, with ring-B precursor (+)-E-5 predominating (isolated in 55% yield).[44]:12-17,57-63[6]:186-188[12][1]:542-543 The isomeric lactam-lactone could be isomerized to (+)-E-5 by treatment in methanolic HCl.[59]:24-26,81-84  Figure 21: ETH synthesis of the B-C-component: Alternative Synthesis of the (racemic) Ring-B Precursor (only one enantiomer shown for racemates) Alternative synthesis of racemický ring-B precursor from Hagemann's ester: implementation of the amidacetal-Claisen rearrangement. Five steps were needed to transform Hagemann's ester rac-E-6 into the racemate of the lactam-lactone rac-E-5 form of the ring-B precursor.[58]:14-31[6]:188-190 The product of the C-methylation step rac-E-6 → rac-E-7 (NaH, CH3Já ) was purified via its crystalline oxim. The cis-hydroxy-ester (configuration secured by lactone formation[58]:64) resulting from the reduction step rac-E-7 → rac-E-8 (NaBH4 ) had to be separated from the trans izomer. The thermal přeskupení rac-E-8 → rac-E-9 představuje implementace z amidacetal-Claisen rearrangement in organic synthesis,[74][58]:36-49 a precedent to Johnson's orthoester-Claisen a Ireland's ester-enolate rearrangement.[75] Ozonolýza (Ó3 /MeOH, HCOOH /H2Ó2 ) z N, N-dimethylamide ester rac-E-9 afforded dilactone acid rac-E-10, from which two reactions led to lactam-lactone methylester rac-E-7, the racemate of ring-B precursor (+)-E-7.[58]:57-67 Determination of absolute configuration of (+)-ring-B precursor via its conversion into the (+)-ring-C precursor  Figure 22: ETH synthesis of the B-C-component: Conversion of Ring-B Precursor to Ring-C Precursor The conversion of ring-B precursor into the ring-C precursor was based on a reductive dekarbonylace z thiolakton E-12 with chloro-tris-(triphenylphosphino)-rhodium(I).[42]:14-32[6]:191-193[12] Treatment of a methanolic solution of ring-B precursor (+)-E-5 with diazomethane in the presence of katalytické amounts of methoxid sodný, followed by thermal odstranění of methanol, gave methylidene lactam E-11, which was converted to the thiolactone E-12 with liquid H2S containing a catalytic amount of trifluoracetic acid.[42]:15-16,56-58 Topení E-12 in toluene with the Rh(I)-complex afforded ring-C precursor (−)-E-13 besides the corresponding cyclopropane derivative E-14. Ring-C precursors prepared via this route and from (+)-camphor at Harvard [1]:540-542 were found to be identical: (−)-E-13 ≡ H-41.[42]:33-34 Ozonolysis of ring-C precursor (−)-E-13 dal sukcinimid derivát (−)-E-15.[42]:33-35,88-89 This succinimide was found to be identical[6]:193[1]:543-544 in constitution and optická rotace (i.e., configuration) with the corresponding succinimide derived from ring C of Vitamin B12, isolated after ozonolysis of crystalline heptamethyl-cobyrinate (cobester[poznámka 9]) prepared from Vitamin B12.[54]:9-18,67-70 The approach pursued at Harvard for conversion of ring-B precursor into ring-C precursor was based on a fotochemické degradation of the acetic acid side chain carboxyl group, starting from (+)-E-7 prepared at ETH.[poznámka 17] Coupling of ring-B and ring-C precursors to the B-C-component. Implementation of the sulfide contraction C,C-condensation method The iminoester /enamine C,C-kondenzace method for constructing the vinylogický amidine system, developed in the model studies on corrin syntéza,[26][33] failed completely in attempts to create the targeted C,C-bond between ring-B precursor (+)-E-5 with ring-C precursor (−)-E-13 to give the B-C-component E-18.[6]:193-194[8]:379[1]:544 The problem was solved by "intramolecularization" of the bond formation process between the elektrofilní (thio)iminoester carbon and the nukleofilní methylidene carbon of the enamin system through first oxidatively connecting these two centers by a sulfur bridge, and then achieving the C,C-bond formation by a now intramolecular thio-iminoester/enamine condensation with concomitant transfer of the sulfur to a thiophile.[6]:194-197[8]:380-386[18]:1537-1538 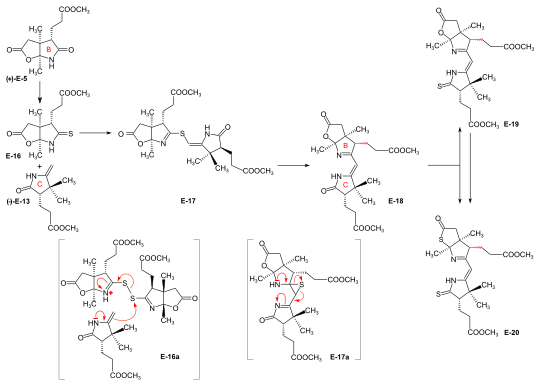 Figure 23: ETH synthesis of the B-C-component: coupling of the ring B and C precursors (implementation of C/C-coupling by the sulfide-contraction method) Conversion of lactam (+)-E-5 into the corresponding thiolactam E-16 (Str2S5),[44]:20-23,74-75 oxidation of E-16 s benzoylperoxid in the presence of ring-C precursor (−)-E-13 (prepared at Harvard by the Cornforth route[poznámka 6]), followed by heating the reaction product E-17 v triethylphosphite (as both solvent and thiophile) afforded B-C-component E-18 as a (not separated) mixture of two epimers (regarding the configuration of the propionic side chain at ring B) in up to 80 % yield.[44]:38-43,96-102[31]:16-19[8]:381-383[46]:20-21,50-52 The bracketed formulae in the reaction scheme illustrate the type of mechanismus operating in the process: E-16a = primary coupling of E-12 a E-10 na E-13; E-17a = extrusion of the sulfur atom (captured by thiophile) to E-14, where it is left open whether this latter process occurs at the stage of the episulfide. This reaction concept developed at this stage, dubbed sulfide contraction,[6]:199[45][18]:1534-1541[35]:1927-1941 turned out to make possible the construction of all three meso-carbon bridges of the vitamin's corrin ligand in both approaches of the synthesis.[12][11][2]:288-292,297-300[3]:158-164 The conversion of bicyclic lactone-lactam E-18 into the corresponding thiolactone-thiolactam E-20 was brought about by heating with P2S5 /4-methylpyridine v xylen at 130 °C; milder condition produced thiolactam-lactone E-19, používá spojka with the Harvard A-D-components.[49]:73-83 |
| Coupling of the B-C-component with ring-D and ring-A precursors |
|---|
Synthesis of ring-D precursor for the A/D approach 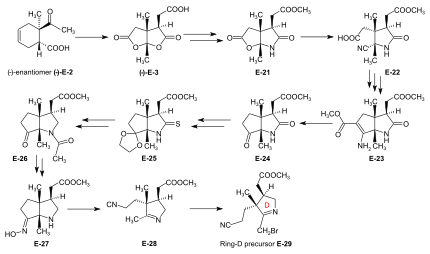 Figure 24: ETH A/D approach to cobyric acid: synthesis of ring-D precursor The starting material for the ring-D precursor,[59]:40-61[61]:17-22[12] the (−)-enantiomer of the dilactone-carboxylic acid (−)-E-3, was prepared from the (−)-enantiomer of the Diels-Alder adukt (−)-E-2[poznámka 18] by oxydation with kyselina chromová /sulfuric acid in acetone.[41]:35,72-73 Léčba (−)-E-3 with NH3 in MeOH gave a lactone-lactam-acid which was esterified with diazomethane to the ester E-21,[59]:104-110 the lactone ring of which was opened with KCN in MeOH to give E-22.[59]:114-116 Conventional conditions of an Arndt-Eistert reaction (SOCl2: acid chloride, then CH2N2 in THF: diazoketone, treated with Ag2Ó in MeOH) led to an – unforeseen, yet useful – ring closure of the originally formed chain-elongated ester through participation of the cyano group as a neighboring elektrofil, affording the bicyclic enamino-ester derivative E-23.[59]:116-120 Hydrolysis with aqueous HCl, accompanied by decarboxylation, and re-esterification with diazomethane gave keto-lactam-ester E-24.[59]:123-126[61]:40-41 Ketalization ((CH2ACH)2, CH (OCH3)3, TsOH ) z E-24 and conversion of this lactam-ester to thiolactam E-25 (P2S5 ) was followed by reductive removal of the sulfur with Raney nikl, acetylace of the amino group, and hydrolysis of the ketal (AcOH) to afford E-26.[61]:42-59 This was converted by deacetylation of the amino group with HCl, and then by treatment with NH2OH/HCl, MeOH/NaOAc into oxime E-27. Beckmann fragmentation (HCl, SOCl2 in CHCl3, N-polystyryl-piperidine) of this oxime E-27 produced imino-nitrile E-28,[61]:60-67 which, when treated with bróm (in MeOH, phosphate nárazník pH 7.5, -10 °C) gave ring-D precursor E-29.[49]:84-88 Conversion of the ring-B precursor into the ring-A precursor for the A/D approach  Figure 25: ETH A/D approach to cobyric acid: conversion of ring-B precursor into ring-A precursor The ring-A precursor (−)-E-31 required in the A/D approach is a close derivative of ring-B precursor (+)-E-5. Its preparation from (+)-E-5 required opening of the lactone group (KCN in MeOH), followed by re-esterification with diazomethane to E-30, then conversion of the lactam group into a thiolactam group with P2S5 poddat se (−)-E-31.[49]:63-72[12] Coupling of the B-C-component with ring-D and ring-A precursors The most efficient way of attaching the two rings D and A to the B-C-component E-18 was to convert E-18 directly into its thiolactam-thiolakton derivát E-20 and then to proceed by first coupling ring-D precursor E-29 to ring C, and then ring-A precursor E-31 to ring B, both by the sulfide contraction method.[49]:26-31[9]:80-83[12] The search for the reaction conditions for these attachments was greatly facilitated by exploratory work done on the two sulfide contraction steps in the A/B approach model study.[49]:27[46]:22-39[2]:285-300  Figure 26: ETH A/D approach to cobyric acid: Attaching ring-C and ring-A precursors to the B-C-component to yield the A/D-seco-corrin Attachment of ring-D precursor E-29 to the ring-C thiolactam in E-20 by sulfide contraction via alkylative coupling (t-BuOK in t-BuOH/THF, tris-(β-cyano-ethyl)-phosphin/CF3COOH v sulfolan ) afforded the B/C/D-sesqui-corrinoid E-32.[49]:89-97 To attach ring-A precursor E-31, the ring B of E-32 was induced to expose its exocyklický methyliden dvojná vazba by treatment with dimethylamine in MeOH (using the method[poznámka 19] developed by Schneider[46]:32-34) forming E-33[49]:108-115 which was subjected to the following cascade of operations:[49]:130-150 iodination (N-jodosukcinimid, CH2Cl2, 0°), coupling with the thiolactam sulfur of the ring-A precursor E-31 [(CH3)3Si]2N-Na in benzene/t-BuOH), complexation (Cd(ClO4)2 in MeOH), treatment with trifenylfosfin /CF3COOH in boiling benzene (sulfide contraction) and, finally, re-complexation with Cd(ClO4)2/N, N-diisopropylethylamine in benzene/MeOH). These six operations, all carried out without isolation of meziprodukty, gave A/D-seco-corrin complex E-34 as mixture of peripheral epimers (separable via HPLC[49]:143-147) in 42-46 % overall yield.[49]:139 |
| A/D-corrin-ring closure by the photochemical A/D-seco-corrin→corrin cycloisomerization |
|---|
A/D-corrin-ring closure by the photochemical A/D-seco-corrin→corrin cycloisomerization to dicyano-cobalt(III)-5,15-bisnor-a,b,d,e,g-pentamethyl-cobyrinate-c-N, N-dimethylamide-f-nitrile (the common corrinoid intermediate) The conditions and prerequisites for the final (A⇒D)-corrin -ring closure were taken over from extensive corrin model studies.[34][76][9]:71-74,83-84[18]:1565-1566[35]:1942-1962 Problems specific to the cobyric acid synthesis that had to be tackled were:[9]:84-88 the possible formation of two diastereomerní A/D-trans-junctions in the ring closure,[49]:37-38 exposure of the methylidene double bond at ring A of the A/D-seco-corrin E-34 in a labile Cd complex,[49]:35-36[18]:1566 and epimerizability of the peripheral stereogenic centers C-3, C-8 and C-13 before and after ring closure.[49]:39[3]:148-150 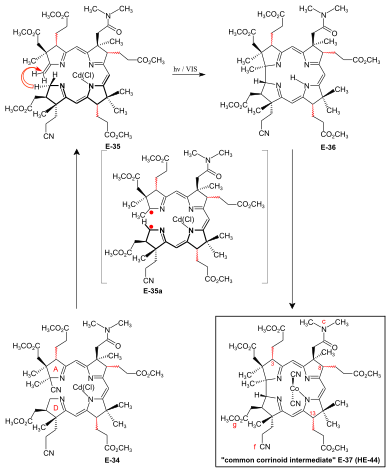 Figure 27: ETH A/D approach to cobyric acid: photochemical A/D-seco-corrin→corrin cycloisomerization to the common corrinoid intermediate In the application of this novel process in the A/D approach of the cobyric acid synthesis,[9]:86-95[49]:39-53[12]:1419 the reaction proceeded most efficiently and with highest cívka stereoselektivita in favor of the natural A/D-trans junction in an A/D-seco-corrin cadmium complex.[49]:42-45[3]:166 Treatment of Cd-complex E-34 as mixture of peripheral epimers s 1,8-Diazabicyclo(5.4.0)undec-7-ene v sulfolan at 60 °C under strict protection against light to odstranit the cyano group at ring A, directly followed by re-treatment with Cd(ClO4)2, led to labile[49]:172 A/D-seco-corrin complex E-35 as a mixture of peripheral epimers. This was directly subjected to the key step, the photochemical ring closure reaction under rigorous exclusion of air:[49]:40 visible light, under Argon, MeOH, AcOH, 60° C. Product of the A/D-ring closure was the free corrin ligand E-36, as the originally formed Cd-corrinate – in contrast to the Cd-seco-corrinate E-35 – decomplexes in the reaction medium.[49]:173[12]:1419 Corrin E-36 was immediately complexed (CoCl2,[18]:1499-1500,1563-64 KCN, air, H2O, CH2Cl2) and finally isolated (thick-layer chromatografie ) as mixture of peripheral epimers in 45-50 % yield over four operations:[49]:169-179 the common corrinoid intermediate dicyano-cobalt(III)-complex E-37 ≡ HE-44.[poznámka 20] HPLC analysis of this mixture E-37 showed the presence of six epimers with natural ligand helicity (Σ 95%, CD spectra ), among them 26% of natural diastereomer 3α,8α,13α, and an equal amount of its C-13 neo-epimer 3α,8α,13β.[49]:46,179-186[12]:1414 Two HPLC fractions (Σ 5%) contained diastereomers with unnatural ligand helicity, as shown by inverse CD spectra.[49]:42-43 Product mixtures from several such cycloisomerizations were combined for preparative HPLC separation and full characterization of the 14 isolated diastereomers of E-37[49]:207-251 (of 16 theoretically possible, regarding helicity and the epimeric centers C-3, C-8, C-13[49]:39).  Figure 28: ETH A/D approach to cobyric acid: coil selectivity in A/D-ring closure In an analytical run, the mixture of cadmium-seco-complex epimers E-35 was separated by HPLC (in the dark) into the natural chloro-cadmium-3α,8α,13α-A/D-seco-corrinate diastereomer (ααα)-E-35 and four other epimer fractions[49]:281-293 Na ozáření[49]:53[12] and following cobaltation, (ααα)-E-35 vyrobeno E-37 in yields of 70-80% as an essentially dual mixture of mainly the 3α,8α,13α epimer, besides some 3α,8α,13β epimer. Less than 1% of fractions with unnatural coil were formed (HPLC, UV/VIS, CD ).[49]:293-300 Mechanistically, fotochemické A/D-seco-corrin corrin cycloisomerization involves an antarafacial sigmatropic shift of the α-hydrogen of the CH2 position C-19 at ring D to the CH2 position of the methylidene group at ring A within a trojice vzrušený stav, creating a transient 15-center-16-electron π-system (see E-35a v obr. 27 ) that antarafacially collapses between positions C-1 and C-19 to the corrin system.[34][35]:1946,1967-1993[77] The coil selectivity of the ring closure in favor of the corrin ligand's natural helicity is interpreted as relating to the difference in steric hindrance between the g-methoxycarbonyl acetic acid chain at ring D and the methylidene region of ring A in the two possible helical coil configurations of the A/D-seco-corrin complex (fig. 28).[49]:38[35]:1960-1962 |
ETH/Harvard: the jointly executed final steps from the common corrinoid intermediate to cobyric acid
The final steps from the common corrinoid intermediate E-37/HE-44 to cobyric acid E-44/HE-51 were carried out by the two groups collaboratively and in parallel, the ETH group working with material produced by the A/D approach a Harvard group with that from the A/B approach.[61]:15[53]:22[55]:47[14]:12[18]:1570-1571 What the two groups in fact accomplished thus were the common final steps of two different syntheses.[11][12]
The tasks in this end phase of the project were the regioselective introduction of methyl groups at the two meso positions C-5 and C-15 of E-37/HE-44, followed by conversion of all its peripheral carboxyl functions do primary amide groups, excepting that in side chain f at ring D, which had to end up as free carboxyl. These conceptually simple finishing steps turned out to be rather complex in execution, including unforeseen pitfalls like a dramatic loss of precious synthetic material in the so-called "Black Friday" (July 9, 1971).[53]:39-40,107-118[9]:97-99[3]:168-169[5](0:07:54-0:09:33)[18]:1568-1569
| Introduction of methyl groups in two meso positions |
|---|
 Figure 29: ETH/Harvard joint final steps: Introduction of methyl groups at the meso positions C-5 and C-15 This introduction of methyl groups could draw on exploratory studies on model corrins[7]:13-14[8]:375-377[78][18]:1528,1530-1532 as well as on exploratory experiments carried out at ETH on cobester[poznámka 9] and its (c→C-8)-lactone derivative.[53]:27-43 Chloromethyl benzyl ether alkylated the meso position C-10 of cobester, but not that of the corresponding lakton, the difference in behavior reflecting the difference in steric hindrance exerted on the meso position C-10 by its neighboring substituents.[53]:37-39 This finding was decisive for the choice of the substrate to be used for introducing methyl groups at meso positions C-5 and C-10 of E-37/HE-44.[9]:96-99[53]:19[3]:167[18]:1567-1568 In this final phase of the synthesis, HPLC again turned out to be absolutely indispensable for separation, isolation, characterization and, above all, identification of pure isomers of dicyano-cobalt(III)-complexes of totally as well as partially synthetic origin.[9]:96-102[3]:165[53]:61-63[5](0:21:13-0:25:28)[18]:1566-1567 The first step was to convert the c-N, N-dimethylcarboxamide group of E-37/HE-44 into the (c→C-8)-lactone derivative E-38/HE-45 by treatment with jód /AcOH effecting iodination at C-8, followed by intramolecular Ó-alkylation of the carboxamide group to an iminium salt that hydrolyses to the lactone.[61]:23,90-108[3]:166-167[4](2:02:18-2:09:02) This lactonization leads to cis-fused rings.[53]:19[5](0:09:34-0:10:43) Reaction of (c→C-8)-lactone E-38/HE-45 with chloromethyl benzyl ether in acetonitril in the presence of LiCl gave, besides mono-adduct, the bis-benzyloxy adduct E-39/HE-46. Při léčbě thiofenol, this produced the bis-phenylthio-derivative E-40/HE-47. Léčba Raney nikl in MeOH not only set free the two methyl groups at the meso positions, but also reductively opened the lactone ring to the free c-carboxyl group at ring B, producing the correct α-konfigurace at C-8. Esterifikace of c-carboxyl with diazomethane afforded hexamethylester-f-nitrile E-41/HE-48.[53]:19-21,39-43,146-205[3]:167-169 For steric reasons, only the predominant[53]:19[61]:24[4](2:08:20-2:09:02) C-3 α-epimer (with the C-3 side chain below the plane of the corrin ring) reacted to a 5,15-disubstituted produkt E-38/H-45, the reaction thus amounting to a chemical separation of the C-3 epimers.[53]:40[5](0:12:51-0:14:33,0:15:56-0:16:24) In improved procedures developed at Harvard later in 1972,[18]:1569 footnote 62 the reagent chloromethyl benzyl ether was replaced by formaldehyd /sulfolane/HCl in acetonitrile for the alkylation step, and Raney nickel in the snížení step was replaced by zinc/acetic acid to give E-41/HE-48.[5](0:00:32-0:21:12) |
| Dicyano-cobalt(III)-3α,8α,13α-a,b,c,d,e,g-hexamethyl-cobyrinate-f-amide: Identification with material derived from vitamin B12 |
|---|
Figure 30: ETH/Harvard joint final steps: hexamethylcobyrinate-f-amide (synthesis and identification) to cobyric acid Concentrated H2TAK4 at room temperature converted the nitrile function of pure (3α,8α,13α)-E-41/HE-48 into the primary f-amide skupina E-42/HE-49, besides partial epimerization at C-13;[9]:100-103[53]:21,134-136[3]:150-151,169-170 an alternative procedure for the selective f-nitrile→f-amide conversion (BF3 in CH3COOH) later developed at Harvard proceeded without epimerizace at C-13.[18]:1569 footnote 62[5](0:46:40-0:49:45)[53]:21 A crystalline sample of the 3α,8α,13α-epimer of dicyano-cobalt (III)-a,b,c,d,e,g-hexamethyl-cobyrinate-f-amide E-42/HE-49, isolated by HPLC, was the first totally synthetic intermediate to be chromatographically and spektroskopicky identified with a relay sample made from vitamin B12.[53]:136-141[3]:170 In the remaining steps of the synthesis, only epimerization at C-13 played an important role,[53]:19-21 with 13α being the configuration of the natural corrinoids, and 13β known as neo-epimers of vitamin B12 and its derivatives;[3]:169-170[79] these are readily separable by HPLC.[5](0:19:30-0:20:21)[53]:135,208-209 In the course of 1972, comprehensive identifications (HPLC, UV/VIS, IR, NMR, CD, mass spectra ) of crystalline samples of totally synthetic intermediates with the corresponding compounds derived from vitamin B12 were carried out in both laboratories: individually compared and identified were the 3α,8α,13α and 3α,8α,13β neo-epimer of f-amide E-42/HE-49, as well as the corresponding pair of C-13-epimeric nitriles E-41/HE-48.[53]:206-221[55]:46-47[5](0:27:28-0:46:32) All these dicyano-cobalt(III)-complexes are soluble in organic solvents[54]:11 in which the separation power of HPLC by far exceeds that of analytical methods operating in water,[53]:44-45 the solvent in which cobyric acid was to be identified, and where it exists as two easily equilibrating aquo-cyano complexes, epimeric regarding the position of the two non-identical axial Co ligandy.[61]:196-197[55]:49-60 These thorough identifications of the totally synthetic with partially synthetic materials mark the accomplishment of the two syntheses. They also reciprocally provided structure proof for a specific constitutional isomer isolated from a mixture of isomeric mono-amides formed in the partial ammonolysis of the B12-derived cobester,[poznámka 9] tentatively assigned to be the 3α,8α,13α-f-amide E-42/HE-49 (see fig. 30).[54]:9-18,67-70[53]:226-239[57] |
| Synthetic cobyric acid |
|---|
The final task of reaching cobyric acid from f-amide E-42/HE-49 required the critical step of hydrolysing the singular amide function into a free carboxyl function without touching any of the six methoxycarbonyl groups around the molecule's periphery. Since exploratory attempts by the conventional method of amide hydrolysis via nitrosace led to detrimental side reactions at the chromofor, a novel way of "hydrolysing " the f-amide group without touching the six methylester groups was conceived and explored at ETH: treatment of f-amide E-42/HE-49 (B12-derived relay material) with the unusual reagent α-chloro-propyl-(N-cyclohexyl)-nitrone[80] a AgBF4 in CH2Cl2, then with HCl in H2Ó/dioxan, and finally with dimethylamine in isopropanol afforded the f-acid E-43/HE-50 in 57% yield.[61]:24-25,159-172[3]:170-172[5](0:53:17-0:58:30) Sustained experimentations at Harvard eventually showed the nitrosation method to be successful (N2Ó4, CCl4, NaOAc ) and to produce the f-carboxyl group even more effectively.[3]:172-173[5](0:58:19-0:59:15) It was also at Harvard that conditions for the last step were explored, conversion of all remaining ester groups into primary amide groups by ammonolysis. Tekutý amoniak v ethylenglykol, in the presence of NH4Cl and the absence of oxygen, converted f-carboxy-hexamethylester E-43/HE-50 into f-carboxy-hexa-amide E-44/HE-51 (= cobyric acid).[3]:173-175[53]:24 This was crystallised and shown both as the α-cyano-β-aquo and the α-aquo-β-cyano form to be chromatographically and spectroscopically identical with the corresponding forms of natural cobyric acid.[5](0:59:53-1:09:58)[3]:175-176[61]:26-27,196-221 At Harvard, the transformation E-43/HE-50 → E-44/HE-51 was eventually carried out starting with f-amide that had been obtained by celková syntéza via the A/B approach.[55]:47-61 The ETH group contented itself with a corresponding f-amide → cobyric acid conversion and subsequent cobyric acid identification where the actual starting material f-amide was derived from vitamin B12.[53]:22[61]:15[12]:footnote 45[18]:1570-1571 |
Poznámky
- ^ For a review about syntheses of corrins, see[25]; this includes more recent synthetic approaches to vitamin B12 by the groups of Stevens,[25]:293-298 Jacobi,[25]:298-300 a Mulzer,[25]:300-301 as well as references to approaches by Todde nebo Cornforth (viz také[43]:261-268) preceding the efforts by Eschenmoser a Woodward.[18]:1493-1496
- ^ A b C d E Formulae in fíky. 4 a 6 illustrate the atom, ring, and side chain enumeration in corrins: "Nomenclature of Corrinoids". Čistá a aplikovaná chemie. 48 (4): 495–502. 1976. doi:10.1351/pac197648040495.
- ^ The year 1964 refers to the first corrin synthesis of a pentamethylcorrin via A/B-cyclization by iminoester/enamine-C,C-condensation;[26] the heptamethylcorrin shown here (M = Co(CN)2) was prepared by the same ring closure method in 1967.[27]
- ^ A b Friedrich, W.; Gross, G.; Bernhauer, K.; Zeller, P. (1960). "Synthesen auf dem Vitamin-B12-Gebiet. 4. Mitteilung. Partialsynthese von Vitamin B12". Helvetica Chimica Acta. 43 (3): 704–712. doi:10.1002/hlca.19600430314. For recent partial syntheses of vitamin B12 a coenzyme B12 from cobyric acid, see Widner, Florian J.; Gstrein, Fabian; Kräutler, Bernhard (2017). "Partial Synthesis of Coenzyme B12 from Cobyric Acid". Helvetica Chimica Acta. 100 (9): e1700170. doi:10.1002/hlca.201700170.
- ^ A b Vidět Determination of absolute configuration of (+)-ring-B precursor via its conversion into the (+)-ring-C precursor in (Show/Hide) "Synthesis of the ETH B-C-component (part of the A/B as well as A/D approach) ".
- ^ A b C d Dopis od J. W. Cornforth to A. Eschenmoser, April 16th, 1984, see [18]:1561 footnote 51; see also refs.[6][42]:40[43]:265. This preparation of a ring-C precursor from (+)-camphor zapojen 8 steps, compared to 4 steps[poznámka 5] from the ETH ring-B precursor (but it used a commonly available precursor instead of "precious" material!)
- ^ A b Vidět Synthesis of the A-D-component carrying the propionic acid function at ring D as methoxycarbonyl group (model A-D-component) in (Show/Hide) "The Harvard synthesis of the A-D-components for the A/B approach ".
- ^ A b Vidět Synthesis of the A-D-component carrying the propionic acid function at ring D as nitrile group in (Show/Hide) "The Harvard synthesis of the A-D-components for the A/B approach ".
- ^ A b C d E Cobester (dicyano-Co-cobyrinic acid heptamethylester) is a non-natural cobyric acid derivative that had played an important subsidiary role in the B12 total syntheses;[53]:14,21,51–90,222–260 it is prepared in one step from vitamin B12 by acid-catalyzed methanolysis.[54]:9–18
- ^ "University of Bristol. WILSON BAKER SYMPOSIUM: Previous Wilson Baker lectures" (PDF). Citováno 2019-10-29.. See also Eschenmoser lecture announcements in "Notizen". Nachrichten aus Chemie und Technik. 20 (5): 89–90. 1972. doi:10.1002/nadc.19720200502..
- ^ A b C Research reports of the Harvard postdoktorandi involved in the vitamin B12 synthesis are in the Harvard archives; vidět "Collection: Papers of Robert Burns Woodward, 1873-1980, 1930-1979 | HOLLIS for Archival Discovery". Citováno 2019-10-29..
- ^ The only "joint publication" is a 1972 interview with Eschenmoser and Woodward in Basle; [29] viz také[18]:1572–1574[62]:1478.
- ^ References given here are a selection from about 50 publications where these epochal syntheses are discussed in more or less detail. Používají se také k výuce syntézy přírodních produktů v pokročilých kurzech nebo na seminářích výzkumných skupin, např. Eschenmoser, A. (2001). "Epilog: Syntéza koenzymu B12: A Vehicle for the Teaching of Organic Synthesis. “In Quinkert, Gerhard; Kisakürek, M. Volkan (eds.). Eseje v současné chemii: Od molekulární struktury k biologii. Curych: Verlag Helvetica Chimica Acta. 391–441. doi:12.1002 / 9783906390451.ch12. ISBN 9783906390284..
- ^ Toto je zatím jediná část příspěvků Harvardu publikovaných s úplnými experimentálními podrobnostmi: Fleming, Iane; Woodward, R. B. (1973). „Syntéza (-) - (R) -trans-β- (l, 2,3-trimethylcyklopent-2-enyl) akrylové kyseliny. Journal of the Chemical Society, Perkin Transactions 1: 1653–1657. doi:10.1039 / P19730001653.Fleming, Iane; Woodward, R. B. (1968). "Exo-2-Hydroxyepicamphor". Journal of the Chemical Society C: Organic: 1289. doi:10.1039 / J39680001289..
- ^ Toto jméno stavebního bloku na levé straně („západní polovina“) se vztahuje k Hesperides, Nymfy Západu, stejně jako Hesperidium a (chemicky zcela nesouvisející) Hesperidin;[1] srov. další barevné názvy od Woodwarda: pentacyclenone,[1]:530 corrnorsteron;[1]:534 korigenolid, korigenát: corrin-genvztyčování seco-corrinů.[2]:285,296 Skupina ETH pojmenovala svůj pravý stavební blok „(thio) dextrolin“ na základě „dexter“, latinsky „right“.[1]:538-539
- ^ Gáforchinon se vyrábí z kafru reakcí s oxid seleničitý: viz White, James D .; Wardrop, Duncan J .; Sundermann, Kurt F. (2002). Zkontrolovali Kenji Koga, Kei Manabe, Christopher E. Neipp a Stephen F. Martin. "Camphorquinone and Camphorquinone Monoxime". Organické syntézy. 79: 125. doi:10.15227 / orgsyn.079.0125..
- ^ A b Wick, Alexander: Report Part I, Harvard University 1967 (nepublikováno[poznámka 11]), citováno v[42]:38–39.
- ^ Vidět Syntézy prekurzoru ring-B v (Zobrazit / Skrýt) "Syntéza ETH B-C složky ".
- ^ Vidět Uzávěr A / B-kroužku v (Zobrazit / Skrýt) "Spojení harvardských A-D komponent s ETH B-C komponentami ".
- ^ Vidět Syntéza dikyano-kobaltu (III) -5,15-bisnor-a, b, d, e, g-pentamethyl-kobyrinátu-c-N, N-dimethylamid-f-nitril (běžný korinoidní meziprodukt) z A-D složky diferencované na kruhu D v (Zobrazit / Skrýt) "Spojení harvardských A-D komponent s ETH B-C komponentami ".
Reference
- ^ A b C d E F G h i j k l m n Ó p q r s t u proti w X y z aa ab ac inzerát ae af ag ah ai aj ak al dopoledne an Woodward, R. B. (1968). „Nedávný pokrok v chemii přírodních produktů“. Čistá a aplikovaná chemie. 17 (3–4): 519–547. doi:10.1351 / pac196817030519.
- ^ A b C d E F G h i j k l m n Ó p q r s t u proti w X y z aa ab ac inzerát ae af ag ah ai Woodward, R. B. (1971). „Nedávný pokrok v chemii přírodních produktů“. Čistá a aplikovaná chemie. 25: 283–304. doi:10.1351 / pac197125010283.
- ^ A b C d E F G h i j k l m n Ó p q r s t u proti w X y z aa ab ac inzerát ae af ag ah ai aj ak al Woodward, R. B. (1973). „Celková syntéza vitaminu B12". Čistá a aplikovaná chemie. 33: 145–178. doi:10.1351 / pac197333010145. PMID 4684454.
- ^ A b C d E F G h i j k l m n Ó p q r s t u proti w Woodward, Robert B. (27. listopadu 1972). R.B. Woodward Celková syntéza vitaminu B12 Přednáška - 1. část (nahraná přednáška). Úvod David Dolphin. Harvard University, Cambridge MA (USA): YouTube. Citováno 2020-01-25.
- ^ A b C d E F G h i j k l m n Ó Woodward, Robert B. (27. listopadu 1972). R.B. Woodward Celková syntéza vitaminu B12 Přednáška - 2. část (nahraná přednáška). Harvard University, Cambridge MA (USA): YouTube. Citováno 2020-01-25.
- ^ A b C d E F G h i j k l m n Ó p Eschenmoser, A. (1968). „Die Synthese von Corrinen“. Moderni Sviluppi della Sintesi Organica (X Corso estivo di chimica, Fondazione Donegani, Frascati 25.9.-5.10.1967) (v němčině). Roma: Accademia Nazionale dei Lincei. 181–214. ISBN 8821804054. ISSN 0515-2216.
- ^ A b C d E F G h i Eschenmoser, A. (1968). "Současné aspekty syntézy korinoidů". Sborník konferencí Nadace Roberta A. Welche o chemickém výzkumu. 12: 9–47. ISSN 0557-1588.
- ^ A b C d E F G h i j k l m n Ó Eschenmoser, A. (1970). „Přednáška k stému výročí (předneseno v listopadu 1969). Cesty do Corrinů“. Čtvrtletní recenze, Chemická společnost. 24 (3): 366–415. doi:10.1039 / qr9702400366.
- ^ A b C d E F G h i j k l m n Eschenmoser, A. (1971). Studie o organické syntéze. XXIII. Mezinárodní kongres čisté a aplikované chemie: speciální přednášky prezentované v Bostonu, USA, 26. - 30. července 1971. 2. London: Butterworths. str. 69–106. doi:10,3929 / ethz-a-010165162. hdl:20.500.11850/84699. ISBN 0-408-70316-4.
- ^ A b C d E F Fuhrer, W .; Schneider, P .; Schilling, W .; Wild, H .; Schreiber, J .; Eschenmoser, A. (1972). „Totalsynthese von Vitamin B12: die photochemische Secocorrin-Corrin-Cycloisomerisierung ". Chimia (abstrakt přednášky). 26: 320.Maag, H .; Obata, N .; Holmes, A .; Schneider, P .; Schilling, W .; Schreiber, J .; Eschenmoser, A. (1972). „Totalsynthese von Vitamin B12: Endstufen ". Chimia (abstrakt přednášky). 26: 320.
- ^ A b C d E F G h i j k l Eschenmoser, A. (1974). „Organische Naturstoffsynthese heute. Vitamin B12 jako Beispiel ". Die Naturwissenschaften. 61 (12): 513–525. Bibcode:1974NW ..... 61..513E. doi:10.1007 / BF00606511. PMID 4453344.
- ^ A b C d E F G h i j k l m n Ó p q r s t u proti w X Eschenmoser, A.; Wintner, C. (1977). „Syntéza přírodních produktů a vitamin B.12". Věda. 196 (4297): 1410–1420. Bibcode:1977Sci ... 196.1410E. doi:10.1126 / science.867037. PMID 867037.
- ^ A b C d E F Zass, E. (2014). "Mezníková celková syntéza dosud nepublikovaná v úplném experimentálním detailu - vitamin B12 (Prezentace přednášek Skolnik Award na 248. národním setkání ACS, San Francisco CA, 12. srpna 2014) ". SlideShare. LinkedIn. Citováno 2020-01-25. Viz také Warr, Wendy (2014). „Herman Skolnik Award Symposium Honouring Engelbert Zass“. Bulletin chemických informací. 66 (4 / zima 2014): 37–40. Citováno 2020-01-25.
- ^ A b C d E F G h Craig, G. Wayne (2016). „Celková syntéza vitaminu B12 - společenství prstenu ". Journal of Porphyrins and Phthalocyanines. 20: 1–20. doi:10.1142 / S1088424615500960.
- ^ A b Nicolaou, K. C.; Sorensen, E. J. (1996). „Kapitola 8: Vitamin B12. R. B. Woodward a A. Eschenmoser (1973) ". Klasika v celkové syntéze: Cíle, strategie, metody. Weinheim: VCH Verlag Chemie. str.99 -136. ISBN 978-3-527-29231-8.
- ^ Marko, I. E. (2001). „Syntéza přírodních produktů: umění totální syntézy“. Věda. 294 (5548): 1842–1843. doi:10.1126 / science.1067545. PMID 11729290.
- ^ A b C d E F G h i j k l m n Eschenmoser, A. (2001). „RBW, vitamin B.12, a Harvard-ETH Collaboration ". In Benfey, O. Theodor; Morris, Peter J. T. (eds.). Robert Burns Woodward - architekt a umělec ve světě molekul. Dějiny série moderních chemických věd. Philadelphia: Nadace chemického dědictví. 23–38. ISBN 978-0941901253. ISSN 1069-2452.
- ^ A b C d E F G h i j k l m n Ó p q r s t u proti w X y z aa ab ac inzerát ae af ag ah ai aj ak al dopoledne an ao ap vod ar tak jako v au av aw sekera ano az ba bb před naším letopočtem bd být bf bg bh bi bj bk bl Eschenmoser, Albert (2015). „Corrinovy syntézy. Část I“. Helvetica Chimica Acta. 98 (11–12): 1483–1600. doi:10.1002 / hlca.201400277.
- ^ A b Nicolaou, K. C.; Sorensen, E. J .; Winssinger, N. (1998). „Umění a věda syntézy organických a přírodních produktů“. Journal of Chemical Education. 75 (10): 1225–1258. Bibcode:1998JChEd..75.1225N. doi:10.1021 / ed075p1225.
- ^ Nicolaou, K. C.; Vourloumis, Dionisios; Winssinger, Nicolas; Baran, Phil S. (2000). „Umění a věda totální syntézy na úsvitu dvacátého prvního století“. Angewandte Chemie International Edition. 39 (1): 44–122. doi:10.1002 / (SICI) 1521-3773 (20000103) 39: 1 <44 :: AID-ANIE44> 3.0.CO; 2-L. PMID 10649349.
- ^ Eschenmoser, Albert (1988). „Vitamin B12: Experimenty týkající se původu jeho molekulární struktury “. Angewandte Chemie International Edition v angličtině. 27: 5–39. doi:10,1002 / anie.198800051.
- ^ Hodgkin, Dorothy Crowfoot; Kamper, Jennifer; MacKay, Maureen; Pickworth, Jenny; Trueblood, Kenneth N.; White, John G. (1956). "Struktura vitaminu B12". Příroda. 178 (4524): 64–66. Bibcode:1956Natur.178 ... 64H. doi:10.1038 / 178064a0. PMID 13348621.
- ^ Glusker, Jenny P. (1995). „Vitamin B12 a B.12 Koenzymy ". Vitamíny a hormony. 50: 1–76.
- ^ A b Bernhauer, K .; Dellweg, H .; Friedrich, W .; Gross, Gisela; Wagner, F. (1960). „Notizen: Vitamin B12-Faktor V1a, ein neuer "inkompletter" Grundkörper der Vitamin B12-Gruppe ". Zeitschrift für Naturforschung B. 15 (5): 336–337. doi:10.1515 / znb-1960-0522.
- ^ A b C d E F Montforts, Franz-Peter; Osmers, Martina; Leupold, Dennis (2012). "Chemická syntéza umělých korinů". V Kadish, Karl M .; Smith, Kevin M .; Guilard, Roger (eds.). Příručka vědy o porfyrinu. 25. World Scientific Publishing. 265–307. doi:10.1142/9789814397605_0020. ISBN 978-981-4397-66-7.
- ^ A b C d Bertele, E .; Boos, H .; Dunitz, J. D.; Elsinger, F .; Eschenmoser, A.; Felner, I .; Gribi, H. P .; Gschwend, H .; Meyer, E. F .; Pesaro, M .; Scheffold, R. (1964). "Syntetická cesta do systému Corrin". Angewandte Chemie International Edition v angličtině. 3 (7): 490–496. doi:10,1002 / anie.196404901.
- ^ Felner-Caboga, I .; Fischli, A .; Wick, A .; Pesaro, M .; Bormann, D .; Winnacker, E.L .; Eschenmoser, A. (1967). "rac.-Dikyano- (1,2,2,7,7,12,12-heptamethylcorrin) -balt (III)". Angewandte Chemie International Edition v angličtině. 6 (10): 864–866. doi:10,1002 / anie.196708643.
- ^ Benfey, O. Theodor; Morris, Peter J. T. (eds.). Robert Burns Woodward - architekt a umělec ve světě molekul. Dějiny série moderních chemických věd. Philadelphia: Nadace chemického dědictví. ISBN 978-0941901253. ISSN 1069-2452.
- ^ A b ""Herr Woodward bedauert, daß die Sache fertig ist. "Woodward und Eschenmoser über Vitamin B12 und die Situation der organischen Chemie ". Nachrichten aus Chemie und Technik. 20 (8): 147–150. 2010. doi:10.1002 / nadc.19720200804.
- ^ A b C d Krieger, J. H. (1973). „Vitamin B12: boj za syntézu “. Chemické a technické novinky. 51 (11 / 12. března): 16–29.
- ^ A b C d E F G h Craig, G. Wayne (2014). „Eschenmoserův přístup k vitaminu B.12 A / D strategií ". Rezonance. 19 (7): 624–640. doi:10.1007 / s12045-014-0064-4.
- ^ Smith, K. M. (1971). "Nedávný vývoj v chemii pyrrolických sloučenin". Čtvrtletní recenze, Chemická společnost. 25: 31. doi:10.1039 / qr9712500031.
- ^ A b C Bertele, Erhard; Scheffold, Rolf; Gschwend, Heinz; Pesaro, Mario; Fischli, Albert; Roth, Martin; Schossig, Jürgen; Eschenmoser, Albert (2015). „Corrinovy syntézy. Část IV“. Helvetica Chimica Acta. 98 (11–12): 1755–1844. doi:10.1002 / hlca.201200342.
- ^ A b C d E F Yamada, Yasuji; Miljkovic, D .; Wehrli, P .; Golding, B .; Löliger, P .; Keese, R .; Müller, K .; Eschenmoser, A. (1969). „Nový typ syntézy Corrinů“. Angewandte Chemie International Edition v angličtině. 8 (5): 343–348. doi:10,1002 / anie.196903431. PMID 4977933.
- ^ A b C d E F G h i j k Yamada, Yasuji; Wehrli, Pius; Miljkovič, Dušan; Wild, Hans-Jakob; Bühler, Niklaus; Götschi, Erwin; Golding, Bernard; Löliger, Peter; Gleason, John; Pace, Brian; Ellis, Larry; Hunkeler, Walter; Schneider, Peter; Fuhrer, Walter; Nordmann, René; Srinivasachar, Kasturi; Keese, Reinhart; Müller, Klaus; Neier, Reinhard; Eschenmoser, Albert (2015). „Corrin Syntheses. Part VI“. Helvetica Chimica Acta. 98 (11–12): 1921–2054. doi:10.1002 / hlca.201500012.
- ^ A b C d Stevens, R. V. (1982). „Celková syntéza vitaminu B12". V Dolphin, D. (vyd.). Vitamin B12. 1. New York: John Wiley & Sons. 169–200. ISBN 978-0-471-03655-5.
- ^ A b C d E Wild, Jost (1964). Synthetische Versuche v Richtung auf natürlich vorkommende Corrinoide (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 3492). doi:10,3929 / ethz-a-000088927. hdl:20.500.11850/132003.
- ^ Woodward, R. B. (1963). "Versuche zur Synthese des Vitamins B12". Angewandte Chemie. 75 (18): 871–872. doi:10,1002 / ange.19630751827.
- ^ A b Woodward, R. B. (1967). "Zachování orbitální symetrie". Aromatičnost. Zvláštní publikace Chemical Society. 21. London: Royal Society of Chemistry. 217–249.
- ^ Peníze, T. (1985). „Gáfor: Chirální výchozí materiál při syntéze přírodních produktů“. Zprávy o přírodních produktech. 2 (3): 253–89. doi:10.1039 / np9850200253. PMID 3906448.
- ^ A b C d E F G Locher, Urs (1964). Darstellung eines Zwischenproduktes zur Synthese von Vitamin B12 (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 3611). doi:10,3929 / ethz-a-000090323. hdl:20.500.11850/131398.
- ^ A b C d E F G h i j k Dubs, Paul (1969). Beiträge zur Synthese von Vitamin B12: Darstellung vinyloger Amidine mit der Sulfidkontraktions-Methode (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4297). doi:10,3929 / ethz-a-000093384. hdl:20.500.11850/133822.
- ^ A b C d Jackson, A. H .; Smith, K. M. (1973). "Celková syntéza pyrrolových pigmentů". V Apsimon, John (ed.). Celková syntéza přírodních produktů. 1. 143–278. doi:10.1002 / 9780470129647.ch3. ISBN 9780471032519.
- ^ A b C d E F G Löliger, Peter (1968). Darstellung eines die Ringe B und C umfassenden Zwischenproduktes zur Synthese von Vitamin B12 (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4074). doi:10,3929 / ethz-a-000093406. hdl:20.500.11850/133844.
- ^ A b C Roth, M .; Dubs, P .; Götschi, E .; Eschenmoser, A. (1971). „Sulfidkontrakce přes alkylační Kupplung: Eine Methode zur Darstellung von β-Dicarbonylderivaten. Über synthetische Methoden, 1. Mitteilung“. Helvetica Chimica Acta. 54 (2): 710–734. doi:10,1002 / hlca.19710540229.
- ^ A b C d E F G h i j k l m n Ó p q r s t u proti w X y z Schneider, Peter (1972). Totalsynthese von Derivaten des Dicyano-cobalt (III) -5,15-bis-nor-cobyrinsäure-hepta-methylesters (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4819). doi:10,3929 / ethz-a-000090603. hdl:20.500.11850/132893.
- ^ Eschenmoser, A. (1994). „B12: vzpomínky a dodatečné myšlenky. “In Chadwick, Derek J.; Ackrill, Kate (eds.). Biosyntéza tetrapyrrolových pigmentů. Ciba Foundation Symposium 180 (Novartis Foundation Symposia 105). Chichester: J. Wiley & Sons. 309–336. ISBN 978-0471939474.
- ^ Eschenmoser, A. (1969). „Role přechodných kovů v chemické syntéze corrinů“. Čistá a aplikovaná chemie. 20 (1): 1–23. doi:10.1351 / pac196920010001.
- ^ A b C d E F G h i j k l m n Ó p q r s t u proti w X y z aa ab ac inzerát ae af ag ah Fuhrer, Walter (1973). Totalsynthese von Vitamin B.12: Der photochemische Weg (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 5158). doi:10,3929 / ethz-a-000086601. hdl:20.500.11850/131362.
- ^ Huber, J. F. K. (1969). "Vysoká účinnost, vysokorychlostní kapalinová chromatografie ve sloupcích". Journal of Chromatographic Science. 7 (2): 85–90. doi:10.1093 / chromsci / 7.2.85.
- ^ Schreiber, J. (1971). „Ein Beispiel zur Anwendung der schnellen Flüssigchromatogarphie in der organischen Synthese“. Chimia. 25 (12): 405–407.
- ^ Hertzog, D. (1973). "Využití de la chromatografie kapalného haute pression na syntetické organizaci". Informace chimique. 119: 229–231.
- ^ A b C d E F G h i j k l m n Ó p q r s t u proti w X y z aa Maag, Hans (1973). Totalsynthese von Vitamin B.12: Dicyano-Co (III) -Cobyrinsäure-Hexamethylester-f-Amid (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 5173). doi:10,3929 / ethz-a-000085446. hdl:20.500.11850/131110.
- ^ A b C d E F Werthemann, Lucius (1968). Untersuchungen an Kobalt (II) - und Kobalt (III) -Komplexen des Cobyrinsäure-heptamethylesters (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4097). doi:10,3929 / ethz-a-000093488. hdl:20.500.11850/133926.
- ^ A b C d E F G h i Woodward, R. B. (1979). „Syntetický vitamin B12". V Zagalak, B .; Friedrich, W. (eds.). Vitamin B12 (Sborník z 3. evropského sympozia o vitaminu B12 and Intrinsic Factor, University of Zurich, 5-8 March, 1979). Berlín: W. de Gruyter. 37–87. ISBN 3-11-007668-3.
- ^ A b Wintner, Claude E. (2006). „Vzpomínka na Ústav organické chemie, ETH Zürich, 1972-1990“. Chimia. 60 (3): 142–148. doi:10.2533/000942906777675029.
- ^ A b Ernst, Ludger; Maag, Hans (2006). „Preparation and Structure Dôkaz of the Four Isomeric Dicyanocobyrinic Acid Hexamethyl Ester Monoamides Nesoucí amidovou skupinu na postranním řetězci kyseliny propionové“. Liebigs Annalen. 1996 (3): 323–326. doi:10.1002 / jlac.199619960306.
- ^ A b C d E F G Wick, Alexander (1964). Untersuchungen in Richtung einer Totalsynthese von Vitamin B12 (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 3617). doi:10,3929 / ethz-a-000090041. hdl:20.500.11850/132537.
- ^ A b C d E F G h i j k Wiederkehr, René (1968). Darstellung von Zwischenprodukten zur Synthese von Vitamin B12 (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4239). doi:10,3929 / ethz-a-000087656. hdl:20.500.11850/131502.
- ^ A b C d Huber, Willy (1969). Beiträge zur Synthese von Vitamin B12: Zum Problem der (C-D) -Verknüpfung (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4298). doi:10,3929 / ethz-a-000090323. hdl:20.500.11850/132700.
- ^ A b C d E F G h i j k l m n Schilling, Walter (1974). Totalsynthese von Vitamin B.12. Darstellung von Zwischenprodukten und partialsynthetische Endstufen (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 5352). doi:10,3929 / ethz-a-000085344. hdl:20.500.11850/131064.
- ^ A b C Eschenmoser, Albert (2015). „Úvodní poznámky k publikační řadě„ Corrin Syntheses-Parts I-VI “'". Helvetica Chimica Acta. 98 (11–12): 1475–1482. doi:10.1002 / hlca.201400399.
- ^ Scheffold, Rolf; Bertele, Erhard; Gschwend, Heinz; Häusermann, Werner; Wehrli, Pius; Huber, Willi; Eschenmoser, Albert (2015). „Corrinovy syntézy. Část II“. Helvetica Chimica Acta. 98 (11–12): 1601–1682. doi:10.1002 / hlca.201200095.
- ^ Pesaro, Mario; Elsinger, Fritz; Boos, Helmut; Felner-Caboga, Ivo; Gribi, Hanspeter; Wick, Alexander; Gschwend, Heinz; Eschenmoser, Albert (2015). „Corrin Syntheses. Part III“. Helvetica Chimica Acta. 98 (11–12): 1683–1754. doi:10.1002 / hlca.201200308.Blaser, Hans-Ulrich; Winnacker, Ernst-Ludwig; Fischli, Albert; Hardegger, Bruno; Bormann, Dieter; Hashimoto, Naoto; Schossig, Jürgen; Keese, Reinhart; Eschenmoser, Albert (2015). „Corrinovy syntézy. Část V“. Helvetica Chimica Acta. 98 (11–12): 1845–1920. doi:10,1002 / hlca.201300064.
- ^ „ETH Research Collection (dříve ETH e-collection)“. ETH Curych. Citováno 2020-01-25.
- ^ Goto, Toshio (1975). "kapitola 11.34: Syntéza vitaminu B12". V Nakanishi, Koji; Goto, Toshio; Sho, Ito; Natori, Shinsaku; Nozoe, Shigeo (eds.). Chemie přírodních produktů. 2. Tokio: Kodansha / Academic Press. 480–496. ISBN 0-12-513902-0.
- ^ Riether, Doris; Mulzer, Johann (2003). „Celková syntéza kyseliny kobyrové: historický vývoj a nedávné syntetické inovace“. European Journal of Organic Chemistry. 2003: 30–45. doi:10.1002 / 1099-0690 (200301) 2003: 1 <30 :: AID-EJOC30> 3.0.CO; 2-I.
- ^ Corey, E. J.; Chow, S. W .; Scherrer, R. A. (1957). „Syntéza α-santalenu a trans-Δ11,12-iso-α-santalen ". Journal of the American Chemical Society. 79 (21): 5773–5777. doi:10.1021 / ja01578a049.Guha, P. C .; Bhattachargya, S. C. (1944). „Série Santalol. II. Syntéza d- a dl-π-hydroxykamfor, d- a dl-teresantalol a d- a dl-tricykloekasantalová kyselina ". Journal of the Indian Chemical Society. 21: 271–280.Corey, E. J.; Ohno, Masaji; Chow, S. W .; Scherrer, Robert A. (1959). "Kyselinou katalyzované štěpení π-substituovaných tricyklenů. Syntéza 3,8-cyklocaforu". Journal of the American Chemical Society. 81 (23): 6305–6309. doi:10.1021 / ja01532a048.Hasselstrom, Torsten (1931). „Studie na derivátech π-kafru. II. Identita kyseliny dihydro-teresantalové s kyselinou 7-π-apokamfan-karboxylovou“. Journal of the American Chemical Society. 53 (3): 1097–1103. doi:10.1021 / ja01354a043.
- ^ Kaski, B. A. (1971). Studie balení do molekulárních krystalů (PhD). Harvardská Univerzita. str. II-1.
- ^ Blaser, Hans-Ulrich (1971). Herstellung und Eigenschaften eines metallfreien Corrin-Derivates (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4662). doi:10,3929 / ethz-a-000091385. hdl:20.500.11850/133210.
- ^ Fischli, Albert (1968). Die Synthese metallfreier Corrine (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4077). doi:10,3929 / ethz-a-000267791. hdl:20.500.11850/137445.
- ^ Jauernig, D .; Rapp, P .; Ruoff, G. (1973). „5-Nor-, 15-Nor- und 5,15-Dinorcorrinoide“. Hoppe-Seyler's Zeitschrift für physiologische Chemie. 354 (8): 957–966.
- ^ Manasse, O .; Samuel, E. (1902). „Reactionen des Campherchinons“. Berichte der Deutschen Chemischen Gesellschaft. 35 (3): 3829–3843. doi:10,1002 / cber.190203503216.
- ^ Wick, A.E .; Felix, Dorothee; Steen, Katharina; Eschenmoser, A. (1964). „Claisen'sche Umlagerungen bei Allyl- und Benzylalkoholen mit Hilfe von Acetalen des N, N-Dimethylacetamidy. Vorläufige Mitteilung ". Helvetica Chimica Acta. 47 (8): 2425–2429. doi:10,1002 / hlca.19640470835.Felix, Dorothee; Gschwend-Steen, Katharina; Wick, A.E .; Eschenmoser, A. (1969). „Claisen'sche Umlagerungen bei Allyl- und Benzylalkoholen mit 1-Dimethylamino-1-methoxy-äthen“. Helvetica Chimica Acta. 52 (4): 1030–1042. doi:10,1002 / hlca.19690520418.
- ^ Johnson, William Summer; Werthemann, Lucius; Bartlett, William R .; Brocksom, Timothy J .; Li, Tsung-Tee; Faulkner, D. John; Petersen, Michael R. (1970). „Jednoduchá stereoselektivní verze Claisenova přesmyku vedoucí k trans-trisubstituovaným olefinickým vazbám. Syntéza skvalenu“. Journal of the American Chemical Society. 92 (3): 741–743. doi:10.1021 / ja00706a074.Irsko, Robert E .; Mueller, Richard H .; Willard, Alvin K. (1976). "Ester enolát Claisenův přesmyk. Stereochemická kontrola prostřednictvím stereoselektivní tvorby enolátu". Journal of the American Chemical Society. 98 (10): 2868–2877. doi:10.1021 / ja00426a033.
- ^ Wild, Hans-Jakob (1972). Die Synthese von Corrin-Komplexen durch photochemische A / D-Cycloisomerisierung (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4848). doi:10,3929 / ethz-a-000090212. hdl:20.500.11850/132648.
- ^ Gardiner, Maureen; Thomson, Andrew J. (1974). "Luminiscenční vlastnosti některých syntetických metalokorinů". Journal of the Chemical Society, Dalton Transactions (8): 820. doi:10.1039 / DT9740000820.
- ^ Winnacker, Ernst-Ludwig (1968). Ligandreaktivität synthetischer Kobalt (III) -Corrin-Komplexe (PDF) (PhD). ETH Zürich (Promotionsarbeit Nr. 4177). doi:10,3929 / ethz-a-000150375. hdl:20.500.11850/136417. Citovat má prázdný neznámý parametr:
|1=(Pomoc) - ^ Bonnett, R .; Godfrey, J. M .; Math, V. B. (1971). „Kyano-13-epikobalamin (Neovitamin B12) a jeho příbuzní “. Journal of the Chemical Society C: Organic. 22: 3736–43. doi:10.1039 / j39710003736. PMID 5167083.
- ^ Kempe, USA; Das Gupta, T. K.; Blatt, K .; Gygax, P .; Felix, Dorothee; Eschenmoser, A. (1972). „α-Chlornitron I: Darstellung und Ag+-induzierte Reaktion mit Olefinen. Über synthetische Methoden, 5. (Vorläufige) Mitteilung ". Helvetica Chimica Acta. 55 (6): 2187–2198. doi:10,1002 / hlca.19720550640.