Selektivní modulátor glukokortikoidových receptorů - Selective glucocorticoid receptor modulator - Wikipedia
| Selektivní modulátor glukokortikoidových receptorů | |
|---|---|
| Třída drog | |
 | |
| Identifikátory třídy | |
| Synonyma | SEGRM; SEGRA; SEGRAM; DIGRA |
| Použití | Potenciálně atopická dermatitida, glaukom, šedý zákal, oční infekce, a další |
| Biologický cíl | Glukokortikoidový receptor |
| Chemická třída | Steroidní; nesteroidní |
| Na Wikidata | |
Selektivní modulátory glukokortikoidových receptorů (SEGRM) a selektivní agonisté glukokortikoidových receptorů (SEGRA) dříve známý jako disociovaných agonistů glukokortikoidových receptorů (DIGRA) jsou skupinou experimentálních léků určených ke sdílení mnoha žádoucích protizánětlivý, imunosupresivní nebo protinádorový vlastnosti klasických glukokortikoid léky, ale s menším počtem vedlejších účinků, jako je atrofie kůže. Ačkoli předklinické důkazy o protizánětlivých účincích SEGRAMů kulminují,[2] v současné době je účinnost těchto SEGRAM na rakovinu do značné míry neznámá.
Selektivní agonisté glukokortikoidových receptorů (SEGRA) jsou historicky a typicky steroidní ve struktuře, zatímco selektivní modulátory glukokortikoidových receptorů (SEGRM) jsou typicky nesteroidní. Kombinovaná zkratka selektivního agonisty a modulátoru glukokortikoidových receptorů je SEGRAM.[2] Byla vyvinuta řada takových ligandů, které jsou hodnoceny v preklinickém a klinickém testování.
SEGRAM dosahuje své selektivity spuštěním pouze podmnožiny glukokortikoidový receptor mechanismy působení.[3][4]
Dějiny
Syntetický steroidy s vlastnostmi podobnými SEGRA byly objeveny již na konci 90. let.[5] Během dvacátých let bylo syntetizováno mnoho potenciálních SEGRAM, většina z nich měla nesteroidní struktury. v in vitro studie na buněčných modelech tyto molekuly SEGRAM se vážou na glukokortikoidový receptor s podobnou afinitou jako dexamethason, silný glukokortikoid, a se schopností potlačovat produkci zánětlivých mediátorů, jako je interleukin 6 a prostaglandin E2.[6] Navíc, in vitro konkrétní SEGRAM může podporovat apoptózu u rakovina prostaty[7] a leukémie.[8]
In vivo studie na myších a potkanech ukázaly, že topicky podávaný SEGRAM inhiboval peroxidáza činnost a formování otok, oba ukazatele protizánětlivé aktivity, srovnatelně s prednisolon. Systémové podávání u myší nebo potkanů naznačuje, že SEGRAM mohou akutně snížit infekce, revmatoidní artritida, astma a kolitida.[2] In vivo v současné době chybí důkazy o tom, zda konkrétní SEGRAM mohou vyvolat podobné účinky než klasický glukokortikoid u nádorových onemocnění. Aktuální preklinické testy ukazují, že dosud dostupné SEGRAM by vyvolaly méně nežádoucích účinků nebo alespoň méně závažných vedlejších účinků než klasické glukokortikoidy.[2] Například atrofie kůže u potkanů byla významně méně výrazná než u prednisolonu ve studii s použitím SEGRAM Mapracorat, a metabolické účinky, jako je přírůstek hmotnosti nebo zvýšení hmotnosti glukóza v krvi prakticky neexistovaly.[9]
Mechanismus účinku

Jak neselektivní glukokortikoidy, tak selektivní agonisté glukokortikoidových receptorů působí vazbou na a aktivují glukokortikoidový receptor (GR). Na rozdíl od glukokortikoidů, které aktivují GR k propracování (alespoň) dvou signální transdukce cesty,[10] SEGRAM aktivuje GR takovým způsobem, že funguje pouze jednou ze dvou hlavních možných cest.[11]
Při absenci glukokortikoidů se GR nachází v cytosol v neaktivním stavu v komplexu s proteiny tepelného šoku (HSP) a imunofiliny. Vazba glukokortikoidů na GR aktivuje receptor tím, že způsobí a konformační změna v GR a tedy disociaci vázaných HSP. Aktivovaný GR pak může regulovat genovou expresi prostřednictvím jedné ze dvou cest:[10]
- Transaktivace
- První (přímá) cesta se nazývá transaktivace přičemž aktivovaný GR dimerizuje, je přemístěn do jádro a váže se na specifické sekvence DNA volala prvky glukokortikoidové odpovědi (GRE). Komplex GR / DNA rekrutuje další proteiny, které přepisují DNA po směru mRNA a nakonec protein. Mezi příklady genů reagujících na glukokortikoidy patří ty, které kódují Annexin A1, TSC22D3 (také známý jako GILZ), enzym konvertující angiotensin, neutrální endopeptidáza a další protizánětlivé proteiny.
- Přepis
- Druhá (nepřímá) cesta se nazývá transreprese, ve kterém je aktivován monomerní GR se váže na další transkripční faktory, jako je NF-kB a AP-1 a brání jim v regulaci exprese jejich cílových genů. Tyto cílové geny kódují proteiny, jako jsou cyklooxygenáza, ŽÁDNÁ syntáza, fosfolipáza A2, faktor nekrózy nádorů, transformující růstový faktor beta, ICAM-1 a řada dalších prozánětlivých proteinů.

Proto jsou protizánětlivé účinky glukokortikoidů výsledkem transaktivace i transreprese. Naproti tomu studie na potkanech a myších ukázaly, že většina vedlejších účinků glukokortikoidů, jako např diabetogenní aktivita, osteoporóza, stejně jako atrofie kůže, jsou způsobeny hlavně transaktivací.[9][12][13] Selektivní glukokortikoid, který je schopen transrepresi bez transaktivace, by měl zachovat mnoho požadovaných terapeutických protizánětlivých účinků a minimalizovat tyto konkrétní nežádoucí vedlejší účinky.[11]
Počáteční důkazy, že samotná transprese může být dostatečná pro protizánětlivou odpověď, byly poskytnuty zavedením a bodová mutace v GR myší, které zabraňovaly GR v dimerizaci a vazbě na DNA, a tím blokování transaktivace.[14][15] Tato mutace zároveň nezasahovala do transreprese. Zatímco GR je nezbytné pro přežití, tyto myši jsou stále životaschopné.[14] Když však byly tyto myši léčeny syntetickým glukokortikoidem dexamethasonem, nedošlo ke zvýšení hladiny glukózy. Tyto myši ošetřené dexamethasonem byly rezistentní na zánětlivý stimul.[15] Tyto myši tedy reagovaly na protizánětlivé účinky dexamethasonu, ale byly rezistentní alespoň k některým vedlejším účinkům.
Stejně jako glukokortikoidy se SEGRAM váže a aktivuje GR. Na rozdíl od glukokortikoidů však SEGRAM selektivně aktivují GR takovým způsobem, že poskytují lepší terapeutický přínos. Obecně platí, že u specifických onemocnění založených na zánětu by SEGRAM měly silněji transrepresivně než transaktivovat, nebo ještě lépe pouze transrepresivně a selhávat v transaktivaci. Tento typ selektivní aktivace GR by měl mít za následek méně nežádoucích účinků než očekávané vedlejší účinky, které se objeví při chronické léčbě klasickými glukokortikoidy.[16]
Klinické testy
Klinické studie fáze II s jednou z kandidátských sloučenin, mapracoratem (kódové názvy BOL-303242-X a ZK 245186[17]), byla zahájena v létě 2009. Jedním z nich byla a dvojitá slepá studie pro zjištění dávky pro mast proti atopické dermatitidě prováděné Intendis, část Bayer HealthCare Pharmaceuticals specializované na dermatologie.[18] Zkušební fáze III byla zahájena v listopadu 2010 a hodnotila oční suspenze pro léčbu zánětu po operaci katarakty provedenou Bausch & Lomb.[19]
Studie fáze II s jiným disociovaným glukokortikoidem fosdagrocorat (PF-04171327) (fosfátový ester proléčivo z dagrocorat (PF-00251802)[20][21]) pro revmatoidní artritidu byla zahájena v roce 2011 Pfizer.[22]
Výsledky těchto klinických studií dosud nebyly zveřejněny a žádný SEGRAM dosud nebyl schválen pro klinické použití.
Potenciální aplikace
U chronických zánětlivých onemocnění, jako je atopická dermatitida (kůže), revmatoidní artritida (klouby), ..., jsou vedlejší účinky kortikosteroidů problematické z důvodu nezbytné dlouhodobé léčby. Proto jsou SEGRAM zkoumány jako alternativní lokální léčba. Systémová dlouhodobá léčba zánětů kortikosteroidy je zvláště náchylná k metabolickým vedlejším účinkům, což činí vývoj orálních SEGRAM zajímavým cílem.[23] Zbývá zjistit, zda selektivní agonisté nebo modulátory receptoru skutečně způsobují v klinických aplikacích významně méně vedlejších účinků než klasické kortikoidy.
Příznivé atrofické účinky
Za zmínku stojí, že atrofické účinky glukokortikoidů nejsou vždy nevýhodou. Léčba hyperproliferativních onemocnění, jako je psoriáza, tuto vlastnost využívá.[24] SEGRAMy by za takových podmínek byly pravděpodobně méně účinné. Nedávné pokroky ukázaly, že první snaha o úplné oddělení GR transreprese a transaktivace pomocí SEGRAM si zaslouží být nuancována, protože protizánětlivé geny stimulované GR transaktivací, jako je GILZ a DUSP1 Zdá se, že hrají důležitou roli.[25][26] Avšak selektivnější povaha těchto SEGRAMů by stále snížila počet vedlejších účinků zprostředkovaných GR a zaslouží si další klinické testování.
Chemie


Brzy SEGRA byly syntetické steroidy. Příkladem je RU 24858, jedna z prvních sloučenin tohoto typu, která má být zveřejněna.[5] Mnoho novějších SEGRA má odlišný rámec, ačkoli podobnost se steroidy lze stále vidět v molekulách, jako je benzopyranochinolin A 276575, nebo v oktahydropenanthren-2,7-diolových derivátech. Ukázalo se, že všechny tyto sloučeniny vykazují vlastnosti SEGRA na buněčných nebo zvířecích modelech.[3]
Mapracorat je jedním z mnoha trifluorpropanolaminů a amidů, které mají méně zjevnou strukturu jako steroidy. Dalšími typickými příklady této skupiny jsou ZK 216348[9] a 55D1E1.[4] Objemné, bicyklické aromatické substituenty (R1 a R.2) vysvětlují strukturální podobnost s kortikoidy. The R konformace z asymetrický uhlík atom se zdá být nezbytný pro afinitu GR.[9]
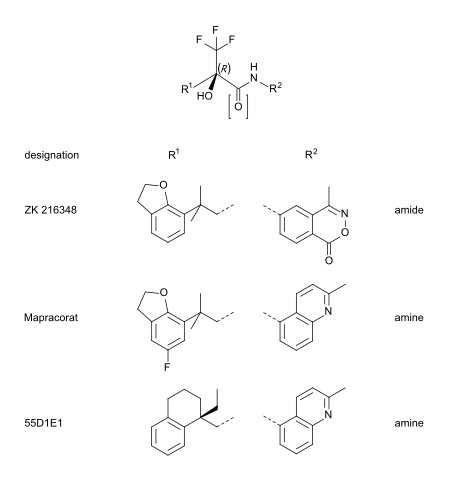
Seznam SEGRM
- Dagrocorat (PF-00251802, PF-251802)
- Fosdagrocorat (PF-04171327, PF-4171327)
- Mapracorat (BOL-303242-X, ZK-245186)
Viz také
- Selektivní modulátor receptoru
- Selektivní modulátor androgenových receptorů
- Selektivní modulátor estrogenových receptorů
- Selektivní modulátor receptoru progesteronu
Reference
- ^ Mealy N; Dulsat C (2009). „36. výroční zasedání Arbeitsgemeinschaft Dermatologische Forschung (ADF)“. Drugs Fut. 34 (4): 341.
- ^ A b C d Sundahl N, Bridelance J, Libert C, De Bosscher K, Beck IM (květen 2015). „Selektivní modulace glukokortikoidových receptorů: Nové směry s nesteroidními lešeními“. Farmakologie a terapeutika. 152: 28–41. doi:10.1016 / j.pharmthera.2015.05.001. PMID 25958032.
- ^ A b C Robinson RP, Buckbinder L, Haugeto AI, McNiff PA, Millham ML, Reese MR, Schaefer JF, Abramov YA, Bordner J, Chantigny YA, Kleinman EF, Laird ER, Morgan BP, Murray JC, Salter ED, Wessel MD, Yocum SA (Březen 2009). „Analogy oktahydrophenanthren-2,7-diolu jako disociované agonisty glukokortikoidových receptorů: objev a zkoumání olova“. Journal of Medicinal Chemistry. 52 (6): 1731–43. doi:10.1021 / jm801512v. PMID 19239259.
- ^ A b Biggadike K, Boudjelal M, Clackers M, Coe DM, Demaine DA, Hardy GW, Humphreys D, Inglis GG, Johnston MJ, Jones HT, House D, Loiseau R, Needham D, Skone PA, Uings I, Veitch G, Weingarten GG , McLay IM, Macdonald SJ (prosinec 2007). „Nesteroidní agonisté glukokortikoidů: tetrahydronaftaleny s alternativními mimetiky steroidních A-kruhů s disociovanou (transrepresí / transaktivací) selektivitou účinnosti“. Journal of Medicinal Chemistry. 50 (26): 6519–34. doi:10.1021 / jm070778w. PMID 18038970.
- ^ A b C Vayssière BM, Dupont S, Choquart A, Petit F, Garcia T, Marchandeau C, Gronemeyer H, Resche-Rigon M (srpen 1997). „Syntetické glukokortikoidy, které disociují transaktivaci a AP-1 transrepresi, vykazují protizánětlivou aktivitu in vivo.“ Molekulární endokrinologie. 11 (9): 1245–55. doi:10.1210 / me.11.9.1245. PMID 9259316.
- ^ A b Lin CW, Nakane M, Stashko M, Falls D, Kuk J, Miller L, Huang R, Tyree C, Miner JN, Rosen J, Kym PR, Coghlan MJ, Carter G, Lane BC (srpen 2002). "trans-aktivační a represivní vlastnosti nového ligandu nesteroidního glukokortikoidového receptoru 2,5-dihydro-9-hydroxy-10-methoxy-2,2,4-trimethyl-5- (1-methylcyklohexen-3-y1) -lH- [1] benzopyrano [3,4-f] chinolin (A276575) a jeho čtyři stereoizomery “. Molekulární farmakologie. 62 (2): 297–303. doi:10,1124 / mol. 62.2.297. PMID 12130681.
- ^ Yemelyanov A, Czwornog J, Gera L, Joshi S, Chatterton RT, Budunova I (červen 2008). „Nová sloučenina fyto-modulátorů steroidních receptorů a inhibuje růst a přežití buněk rakoviny prostaty“. Výzkum rakoviny. 68 (12): 4763–73. doi:10.1158 / 0008-5472.CAN-07-6104. PMID 18559523.
- ^ Lesovaya EA, Yemelyanov AY, Kirsanov KI, Yakubovskaya MG, Budunova IV (listopad 2011). „Protinádorový účinek ligandu nesteroidního glukokortikoidového receptoru CpdA na buněčné linie leukémie CEM a K562“ (PDF). Biochemie. Biokhimiia. 76 (11): 1242–52. doi:10.1134 / S000629791111006X. PMID 22117551. S2CID 35234449.
- ^ A b C d Schäcke H, Schottelius A, Döcke WD, Strehlke P, Jaroch S, Schmees N, Rehwinkel H, Hennekes H, Asadullah K (leden 2004). „Disociace transaktivace od transreprese selektivním agonistou glukokortikoidového receptoru vede k oddělení terapeutických účinků od vedlejších účinků“. Sborník Národní akademie věd Spojených států amerických. 101 (1): 227–32. Bibcode:2004PNAS..101..227S. doi:10.1073 / pnas.0300372101. PMC 314167. PMID 14694204.
- ^ A b Rhen T, Cidlowski JA (říjen 2005). „Protizánětlivý účinek glukokortikoidů - nové mechanismy pro staré léky“. The New England Journal of Medicine. 353 (16): 1711–23. doi:10.1056 / NEJMra050541. PMID 16236742.
- ^ A b Newton R, Holden NS (říjen 2007). „Oddělovací transreprese a transaktivace: nepříjemný rozvod pro glukokortikoidový receptor?“. Molekulární farmakologie. 72 (4): 799–809. doi:10,1124 / mol. 107,038794. PMID 17622575. S2CID 52803631.
- ^ Heinemann A, Schuligoi R (2008). „Glukokortikoid - silný a umstritten“ [Glukokortikoidy - silné a kontroverzní]. Österreichische Apothekerzeitung (v němčině). 62 (23).
- ^ Coghlan MJ, Jacobson PB, Lane B, Nakane M, Lin CW, Elmore SW, Kym PR, Luly JR, Carter GW, Turner R, Tyree CM, Hu J, Elgort M, Rosen J, Miner JN (květen 2003). „Nový protizánětlivý prostředek udržuje účinnost glukokortikoidů se sníženými vedlejšími účinky“. Molekulární endokrinologie. 17 (5): 860–9. doi:10.1210 / me.2002-0355. PMID 12586843.
- ^ A b Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P, Schütz G (květen 1998). „Vazba glukokortikoidového receptoru na DNA není pro přežití nezbytná“. Buňka. 93 (4): 531–41. doi:10.1016 / S0092-8674 (00) 81183-6. PMID 9604929. S2CID 6524157.
- ^ A b Reichardt HM, Tronche F, Bauer A, Schütz G (2000). "Molekulárně genetická analýza signalizace glukokortikoidů pomocí systému Cre / loxP". Biologická chemie. 381 (9–10): 961–4. doi:10.1515 / BC.2000.118. PMID 11076028. S2CID 37837380.
- ^ Schäcke H, Berger M, Rehwinkel H, Asadullah K (září 2007). „Selektivní agonisté glukokortikoidových receptorů (SEGRA): nové ligandy se zlepšeným terapeutickým indexem“ (PDF). Molekulární a buněčná endokrinologie. 275 (1–2): 109–17. doi:10.1016 / j.mce.2007.05.014. PMID 17630119. S2CID 46020217.
- ^ Cavet ME, Harrington KL, Ward KW, Zhang JZ (2010). „Mapracorat, nový selektivní agonista glukokortikoidových receptorů, inhibuje hyperosmolárně indukované uvolňování cytokinů a dráhy MAPK v lidských epiteliálních buňkách rohovky“. Molekulární vidění. 16: 1791–800. PMC 2932489. PMID 20824100.
- ^ Číslo klinického hodnocení NCT00944632 pro „Eskalace dávky různých koncentrací ZK 245186 při atopické dermatitidě“ na ClinicalTrials.gov
- ^ Číslo klinického hodnocení NCT01230125 pro „Oční suspenze Mapracorat pro léčbu zánětu oka po operaci katarakty“ na ClinicalTrials.gov
- ^ Stock T, Fleishaker D, Mukherjee, A, Le V, Xu J, Zeiher B (2009). „Hodnocení bezpečnosti, farmakokinetiky a farmakodynamiky selektivního modulátoru glukokortikoidových receptorů (SGRM) u zdravých dobrovolníků“. Artritida Rheum. 60 (Suppl 10): 420. doi:10,1002 / článek 25503 (neaktivní 9. 11. 2020).CS1 maint: více jmen: seznam autorů (odkaz) CS1 maint: DOI neaktivní od listopadu 2020 (odkaz)
- ^ Hu X, Du S, Tunca C, Braden T, Long KR, Lee J, Webb EG, Dietz JD, Hummert S, Rouw S, Hegde SG, Webber RK, Obukowicz MG (srpen 2011). „Antagonisté, ale ne částeční agonisté ligandů glukokortikoidových receptorů, vykazují podstatnou disociaci vedlejších účinků.“ Endokrinologie. 152 (8): 3123–34. doi:10.1210 / cs.2010-1447. PMID 21558312.
- ^ Číslo klinického hodnocení NCT01393639 pro „Studie porovnávající dávky experimentální glukokortikoidové sloučeniny s prednisonem a placebem při revmatoidní artritidě“ na ClinicalTrials.gov
- ^ Schäcke H, Hennekes H, Schottelius A, Jaroch S, Lehmann M, Schmees N, Rehwinkel H, Asadullah K (2002). „SEGRA: nová třída protizánětlivých sloučenin“. Workshop Ernst Schering Research Foundation (40): 357–71. doi:10.1007/978-3-662-04660-9_20. ISBN 978-3-662-04662-3. PMID 12355726.
- ^ Kerscher MJ, Hart H, Korting HC, Stalleicken D (duben 1995). „In vivo hodnocení atrofogenní účinnosti mometason-furoátu, nově vyvinutého chlorovaného silného topického glukokortikoidu ve srovnání s jinými topickými glukokortikoidy starými i novými“. International Journal of Clinical Pharmacology and Therapeutics. 33 (4): 187–9. PMID 7620686.
- ^ Shah S, King EM, Chandrasekhar A, Newton R (květen 2014). „Role pro mitogenem aktivovanou proteinkinázu (MAPK) fosfatázu, DUSP1, při zpětné vazbě na expresi a represi zánětlivých genů dexamethasonem“. The Journal of Biological Chemistry. 289 (19): 13667–79. doi:10,1074 / jbc.M113.540799. PMC 4036371. PMID 24692548.
- ^ Ayroldi E, Macchiarulo A, Riccardi C (prosinec 2014). „Cílení na vedlejší účinky glukokortikoidů: selektivní modulátor receptorů glukokortikoidů nebo leucinový zip indukovaný glukokortikoidy? Perspektiva“. FASEB Journal. 28 (12): 5055–70. doi:10.1096 / fj.14-254755. PMID 25205742. S2CID 40874311.