Cockaynův syndrom - Cockayne syndrome
| Cockaynův syndrom | |
|---|---|
| Ostatní jména | Neill-Dingwallův syndrom |
| Specialita | Lékařská genetika, neurologie, dermatologie |
Cockaynův syndrom (CS), také zvaný Neill-Dingwallův syndrom, je vzácný a smrtelný autosomální recesivní neurodegenerativní porucha charakterizovaná selháním růstu, zhoršeným vývojem nervový systém, abnormální citlivost na sluneční světlo (fotocitlivost ), oční poruchy a předčasné stárnutí.[1][2][3] Neschopnost prospívat a neurologické poruchy jsou kritériem pro diagnózu, zatímco fotocitlivost, ztráta sluchu, oční abnormality a dutiny jsou další velmi časté rysy.[3] Problémy s některými nebo se všemi vnitřními orgány jsou možné. Je spojena se skupinou poruch zvaných leukodystrofie, což jsou stavy charakterizované degradací neurologické bílá hmota. Základní poruchou je porucha v Oprava DNA mechanismus.[4] Na rozdíl od jiných defektů opravy DNA nejsou pacienti s CS náchylní k rakovině nebo infekci.[5] Cockayneův syndrom je vzácné, ale destruktivní onemocnění, které obvykle vede k úmrtí v první nebo druhé dekádě života. Mutace specifických genů u Cockayneova syndromu je známá, ale rozšířené účinky a jejich vztah s opravou DNA je třeba ještě dobře pochopit.[5]
Je pojmenována po anglickém lékaři Edward Alfred Cockayne (1880–1956), který ji poprvé popsal v roce 1936 a znovu popsal v roce 1946.[6] Syndrom Neill-Dingwall byl pojmenován po Mary M. Dingwall a Catherine A. Neill.[6] Tito dva vědci popsali případ dvou bratrů s Cockaynovým syndromem a tvrdili, že jde o stejnou nemoc, jakou popsal Cockayne. Ve svém článku tito dva přispěli k známkám onemocnění objevením kalcifikací v mozku. Rovněž srovnávali Cockaynův syndrom s tím, co je nyní známé jako Hutchinson – Gilfordův syndrom progerie (HGPS), poté nazývaného progerie, kvůli pokročilému stárnutí, které charakterizuje obě poruchy.[6]
Typy
- CS typu I, „klasická“ forma, je charakterizována normálním růstem plodu s nástupem abnormalit v prvních dvou letech života. Zrak a sluch postupně klesají.[7] Centrální a periferní nervový systém postupně degeneruje až do smrti v první nebo druhé dekádě života v důsledku vážné neurologické degradace. Kortikální atrofie je u CS typu I méně závažná.[8]
- CS typu II je přítomen od narození (kongenitální ) a je mnohem závažnější než CS Type 1.[7] Po narození zahrnuje velmi malý neurologický vývoj. Smrt obvykle nastává do sedmi let. Tento specifický typ byl také označen jako cerebro-okulo-facio-skeletální (COFS) syndrom nebo Pena-Shokeirův syndrom typu II.[7] Syndrom COFS je pojmenován podle účinků, které má na mozek, oči, obličej a kosterní systém, protože onemocnění často způsobuje atrofii mozku, šedý zákal, ztrátu tuku v obličeji a osteoporózu. Syndrom COFS lze dále rozdělit na několik stavů (COFS typy 1, 2, 3 (spojené s xeroderma pigmentosum ) a 4).[9] U pacientů s touto časnou formou poruchy se obvykle projevuje závažnější poškození mozku, včetně snížené myelinizace bílé hmoty, a rozšířenější kalcifikace, včetně mozkové kůry a bazálních ganglií.[8]
- CS typu III, charakterizovaný pozdním nástupem, je obvykle mírnější než typy I a II.[7] Pacienti s typem III se často dožijí dospělosti.
- Syndrom Xeroderma pigmentosum-Cockayne (XP-CS) se vyskytuje, když jedinec také trpí xeroderma pigmentosum, další nemocí na opravu DNA. Některé příznaky každé nemoci jsou vyjádřeny. Například jsou přítomny pihy a abnormality pigmentu charakteristické pro XP. Je vidět neurologická porucha, spasticita a nedostatečný vývoj pohlavních orgánů charakteristických pro CS. Hypomyelinace a obličejové rysy typických pacientů s CS však nejsou přítomny.[10]
Příčiny
Li hyperoxie nebo přebytek kyslík se vyskytuje v našem těle, našem buněčný metabolismus produkují několik vysoce reaktivních forem kyslíku zvaných volné radikály. To může způsobit oxidační poškození buněčných složek včetně DNA. V normálních buňkách naše tělo opravuje poškozené úseky. V případě tohoto onemocnění kvůli drobným vadám v transkripce, dětské genetický zařízení pro syntézu bílkoviny potřebné pro tělo nefunguje na normální kapacitu. To znamená, že vědci věřili, že genetický aparát těchto dětí pro syntézu bílkovin potřebných pro tělo nefunguje normálně. Postupem času tato teorie vyústila v vývojový selhání a smrt. Tělo každou minutu pumpuje 10 až 20 litrů kyslíku krev a přenesl jej do miliard buněk v našich tělech. V normálu molekulární forma, kyslík je neškodný. Nicméně mobilní metabolismus zahrnující kyslík může generovat několik vysoce reaktivních volných radikálů. Tyto volné radikály mohou způsobit oxidační poškození na buněčné složky včetně DNA. V průměru lidská buňka, několik tisíc léze vyskytují se v DNA každý den. Mnoho z těchto lézí je výsledkem oxidační poškození. Každá léze - poškozená část DNA - musí být vyříznuta a DNA opravena, aby byla zachována její normální funkce. Neopravená DNA může ztratit schopnost kódovat bílkoviny. Výsledkem mohou být také mutace. Tyto mutace mohou aktivovat onkogeny nebo umlčet geny potlačující nádory. Podle výzkumu není oxidační poškození aktivních genů přednostně opravováno a v nejzávažnějších případech je oprava zpomalena po celou dobu genom. Výsledná akumulace oxidačního poškození by mohla narušit normální funkce DNA a může dokonce vést ke spuštění programu buněčné smrti (apoptózy). Děti s tímto onemocněním neopravují aktivní geny, kde dochází k oxidačnímu poškození. Oprava oxidačního poškození je normálně rychlejší v aktivních genech (které tvoří méně než pět procent genomu) než v neaktivních oblastech DNA. Výsledná akumulace oxidačního poškození by mohla narušit normální funkce DNA a může dokonce vést ke spuštění programu buněčné smrti (apoptóza ).[Citace je zapotřebí ]
Genetika
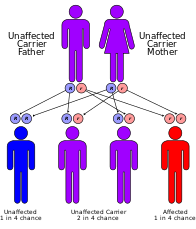
Cockaynův syndrom je klasifikován geneticky jak následuje:
| Typ | OMIM | Gen |
|---|---|---|
| A | 216400 | ERCC8 (také nazývané CSA) |
| B | 133540 | ERCC6 (také nazývaný CSB) |
| C | 216411 | není známo |
- Mutace v genu ERCC8 (také známém jako CSA) nebo v genu ERCC6 (také známém jako CSB) jsou příčinou Cockaynova syndromu.[7] Mutace v mutaci genu ERCC6 tvoří ~ 70% případů. Proteiny produkované těmito geny se podílejí na opravě poškozené DNA prostřednictvím transkripční vázaný opravný mechanismus zejména DNA v aktivních genech. Poškození DNA je způsobeno ultrafialovými paprsky ze slunečního záření, záření nebo volných radikálů v těle. Normální buňka může opravit poškození DNA dříve, než se hromadí. Pokud dojde ke změně genu ERCC6 nebo ERCC8 (jako u Cockaynova syndromu), poškození DNA, ke kterému došlo během transkripce, se neopraví, což způsobí, že se na tomto místě zastaví RNA polymeráza, což interferuje s expresí genu. Jak se hromadí neopravené poškození DNA, brání se postupně aktivnější genové exprese, což vede k nefunkčním buňkám nebo buněčné smrti, což pravděpodobně přispívá k projevům Cockaynova syndromu, jako je předčasné stárnutí a neuronální hypomyelinace.[7]
Mechanismus
Na rozdíl od buněk s normální schopností opravy CSA a CSB deficitní buňky nejsou schopny přednostně opravit cyklobutan pyrimidinové dimery indukované působením ultrafialového (UV) světla na pramen šablony aktivně přepsal geny.[11] Tento nedostatek odráží ztrátu schopnosti provádět proces opravy DNA známý jako transkripční vazba oprava nukleotidové excize (TC-NER).[Citace je zapotřebí ]
V poškozené buňce se protein CSA normálně lokalizuje do míst Poškození DNA, zejména mezivláknové příčné vazby, dvouvláknové zlomy a některé monoadukty.[12] Protein CSB se také běžně získává na poškozená místa DNA a jeho nábor je nejrychlejší a nejsilnější následovně: mezivláknové síťování> dvouřetězcové zlomy> monoadukty> oxidační poškození.[12] Protein CSB tvoří komplex s dalším proteinem pro opravu DNA, SNM1A (DCLRE1A ), 5 '- 3' exonukleáza, který lokalizuje křížové vazby mezi vlákny způsobem závislým na transkripci.[13] Akumulace proteinu CSB v místech dvouřetězcových zlomů DNA probíhá způsobem závislým na transkripci a usnadňuje jej homologní rekombinace oprava zlomů.[14] Během G0 /Fáze G1 buněčného cyklu může poškození DNA spustit rekombinační opravný proces závislý na CSB, který využívá RNA (spíše než DNA ) šablona.[15]
Vlastnosti předčasného stárnutí CS jsou pravděpodobně, alespoň zčásti, způsobeny nedostatky v Oprava DNA (vidět Teorie poškození DNA stárnutí ).[Citace je zapotřebí ]
Diagnóza
Lidé s tímto syndromem mají menší než normální velikost hlavy (mikrocefalie ), jsou malého vzrůstu (nanismus ), jejich oči vypadají zapadlé a mají „starý“ vzhled. Často mají dlouhé končetiny společné kontraktury (neschopnost uvolnit sval v kloubu), shrbený záda (kyfóza ) a mohou být velmi tenké (kazetický ), kvůli ztrátě podkožního tuku. Jejich malá brada, velké uši a špičatý tenký nos často dávají letitý vzhled.[8]Často je také postižena kůže pacientů s Cockaynovým syndromem: hyperpigmentace, křečové nebo pavoučí žíly (telangiektázie ),[8] a vážná citlivost na sluneční světlo jsou běžné, dokonce iu jedinců bez XP-CS. Pacienti s Cockayneovým syndromem často velmi pálí nebo puchýřují při velmi malé expozici teplu. Oči pacientů mohou být ovlivněny různými způsoby a oční abnormality jsou u CS běžné. Šedý zákal a zakalení rohovky (neprůhlednost rohovky ) jsou běžné. Může dojít ke ztrátě a poškození nervů zrakového nervu, což způsobí atrofii zraku.[3] Nystagmus nebo nedobrovolný pohyb očí a žáci, kteří se neroztahují, prokazují ztrátu kontroly nad dobrovolným a nedobrovolným pohybem svalů.[8] Typickým znakem je také pigmentace sítnice a pepře. Diagnóza je stanovena specifickým testem na opravu DNA, který měří regeneraci RNA po vystavení UV záření. Přesto, že je spojován s geny zapojenými do oprava nukleotidové excize (NER), na rozdíl od xeroderma pigmentosum „CS není spojena se zvýšeným rizikem rakoviny.[5]
Laboratorní studie
U pacientů s Cockayneovým syndromem vykazují UV ozářené buňky sníženou syntézu DNA a RNA.https://emedicine.medscape.com/article/1115866-workup#c5 Laboratorní studie jsou užitečné zejména k eliminaci dalších poruch. Například kostní rentgenografie, endokrinologické testy a studie chromozomálních zlomenin mohou pomoci při vyloučení poruch zahrnutých do diferenciální diagnostiky.[Citace je zapotřebí ]
Zobrazovací studie
CT mozku u pacientů s Cockayneovým syndromem může odhalit kalcifikace a kortikální atrofii.[Citace je zapotřebí ]
Další testy
Prenatální hodnocení je možné. Kultivace buněk plodové vody se používá k prokázání nedostatku plodových buněk v syntéze RNA po UV záření.
Neurologie
Zobrazovací studie odhalují rozsáhlou nepřítomnost myelinových obalů neuronů v bílé hmotě mozku a obecnou atrofii kůry.[5] Kalcifikace byly také nalezeny v putamen, oblast přední mozek který reguluje pohyby a pomáhá při některých formách učení,[8] spolu s kůrou.[6] Dále atrofie centrální oblasti mozeček u pacientů s Cockayneovým syndromem může také vést k nedostatečné kontrole svalů, zvláště nedobrovolné, a špatnému držení těla, které je obvykle vidět.[Citace je zapotřebí ]
Léčba
Na tento syndrom neexistuje trvalá léčba, i když pacienti mohou být léčeni symptomaticky. Léčba obvykle zahrnuje fyzikální terapii a drobné operace postižených orgánů, jako je odstranění katarakty.[3] Doporučuje se také nosit opalovací krém s vysokým faktorem a ochranný oděv, protože pacienti se syndromem Cockayne jsou velmi citliví na UV záření.[16] Optimální výživa může také pomoci. Doporučuje se genetické poradenství pro rodiče, protože porucha má 25% pravděpodobnost přenosu na jakékoli budoucí děti a je také možné prenatální testování.[3] Dalším důležitým aspektem je prevence recidivy CS u ostatních sourozenců. Identifikace příslušných genových defektů umožňuje rodičům, kteří již mají jedno postižené dítě, nabídnout genetické poradenství a prenataldiagnostické testování.[17]
Prognóza
The prognóza u pacientů s Cockayneovým syndromem je špatná, protože smrt obvykle nastává ve věku 12 let. [18]Prognóza Cockaynova syndromu se liší podle typu onemocnění. Existují tři typy Cockaynova syndromu podle závažnosti a nástupu příznaků. Rozdíly mezi typy však nejsou vždy jednoznačné a někteří vědci se domnívají, že příznaky a příznaky odrážejí spektrum namísto odlišných typů: Cockayneův syndrom typu A (CSA) se vyznačuje normálním vývojem, dokud dítě není ve věku 1 nebo 2 let starý, kdy se růst zpomalí a všimnete si vývojových zpoždění. Příznaky jsou patrné až po 1 roce. Očekávaná délka života u typu A je přibližně 10 až 20 let. Tyto příznaky se projevují u dětí s CS typu 1. Cockayneův syndrom typu B (CSB), známý také jako „cerebro-oculo-facio-skeletal (COFS) syndrom“ (neboli „Pena-Shokeirův syndrom typu B“), je nejtěžší podtyp. Příznaky jsou přítomny při narození a normální vývoj mozku se zastaví po narození. Průměrná délka života dětí s typem B je do 7 let. Tyto příznaky se projevují u dětí s CS typu 2. Kokayneův syndrom typu C (CSC) se objevuje později v dětství s mírnějšími příznaky než u jiných typů a pomalejší progresí poruchy. Lidé s tímto typem Cockaynova syndromu se dožívají dospělosti s průměrnou délkou života 40 až 50 let. Tyto příznaky jsou patrné u CS typu 3.[Citace je zapotřebí ]
Epidemiologie
Cockayneův syndrom je celosvětově vzácný. U Cockaynova syndromu není hlášena žádná rasová záliba. U Cockaynova syndromu není popsána žádná sexuální záliba; poměr mužů a žen je stejný. Cockayneův syndrom I (CS-A) se projevuje v dětství. Cockayneův syndrom II (CS-B) se projevuje při narození nebo v kojeneckém věku a má horší prognózu.[Citace je zapotřebí ]
Nedávný výzkum
Nedávný výzkum z ledna 2018 zmiňuje různé rysy CS, které jsou globálně vnímány se podobnostmi a rozdíly: CS má výskyt 1 z 250 000 živě narozených a prevalenci 2,5 na milion, což je pozoruhodně konzistentní v různých regionech po celém světě:[19]
| Dotčené části | Klinické příznaky | patologie |
|---|---|---|
| Tvář | Zkrácené tváře. Vpadlé oči, velké uši, hubený špičatý nos. Malá brada. Zubní kaz, hypoplázie skloviny | |
| Kůže, vlasy, hřebíky | Fotocitlivost. Vrásčitá a stárnoucí kůže. Tenké suché vlasy, předčasně šedé vlasy. Chudý žilní přístup. | |
| Centrální nervový systém | Mikrocefalie obvykle začíná ve věku 2 let. Mentální retardace s nízké IQ. Opožděné milníky.Třes, ataxie, záchvaty, tahy, a subdurální krvácení. | Demyelinizace - je nerovnoměrný a segmentový - “Metachromatická leukodystrofie ". Oba oligodendroglia a Schwannovy buňky jsou ovlivněny. Ovlivňuje intelektuální bílá hmota, corpus callosum, mozkový kmen, mícha, a periferní nervy. Neuronální ztráta na více místech, zejména na internetu mozeček. Ztráta buňky předního rohu kvůli anterográdní a / nebo retrográdní degenerace. Kalcifikace [55–95%] z mozková kůra (zejména hloubky sulci, bazální ganglia mozeček, thalamus; také z tepny, arterioly, a kapiláry. Cévní Změny - Řetězová plavidla, zejména v oblastech metachromatické leukodystrofie, kalcifikace v leptomeningeal plavidla, zrychlený ateroskleróza a arterioloskleróza.Glióza je přítomen. Astrocyty a mikroglie může vykazovat nepravidelnost cytoplazma, více jádra. Může být viděn jako vysoce intenzivní bílá hmota FLAIR MRI signály sekvence. Žádný hlavní mozek malformace. Je vidět relativní šetření mozkové kůry, mírné ztenčení kortikální pásky. Normální gyral vzor s rozšířením sulci. Laminování, velikost neuronů a konfigurace neokortex jsou zachovány. Může ukázat temenní okcipitální nadvláda. těžký mozeček atrofie. Ztráta Purkinje, granulární neurony, a v některých případech neurony v zubaté jádro. Dendrity z Purkyňovy buňky mohou být hrubě zdeformované („kaktusové květy“), ferruginované dendrity. Dendrity mají méně větví vyššího řádu. Purkinje “axonální torpéda „Může být přítomen.Ventrikulární zvětšení, zvětšení cisterna magna jsou vidět. Amyloidové plaky, neurofibrilární spleti, Hirano těla není běžně vidět, ačkoli ubikvitin reaktivita z axony současnost, dárek |
| Sluch a vestibulární systémy | Senzorineurální, vysoký tón ztráta sluchu [60–90%]. Smíšené vodivé a senzorineurální ztráta sluchu (44%) Nejčastěji bilaterální, zřídka jednostranný | Ztráta vláskových buněk v kochlea, zejména v bazální obrat. Ztráta neuronů v spirální ganglion. Atrofie sluchové dráhy. Scala communis, zahuštěný sponky kurare, rozšířil prototympanum. Ztráta vláskových buněk v pars superior. Ztráta neuronů v vestibulární ganglion. Zhroucení endolymfatický kanál pars inferior |
| Vidění | Zakalení rohovky. Šedý zákal [36–86%]. Obvykle bilaterální, většina se vyvíjí do 4 let.Pigmentární retinopatie („Sůl a pepř“) [43–89%]. Miotický žáci, Optický disk bledost, Enophthalmos, Úzký oční štěrbiny. | Patchy ztráta melanin pigment granule. Lipofuscin depozice, velké buňky s obsahem pigmentu v a perivaskulární rozdělení. Sítnice pigment epiteliální atrofie a hyperplazie. Ztráta buněk v ganglion a vnější vrstvy jaderných buněk. Vnější i vnitřní segment fotoreceptory jsou ovlivněny. Zrakový nerv atrofie, s částečným demyelinizace, axonální ztráta a glióza |
| Muskuloskeletální systém | Kachektické nanismus. Smlouvy. Kyfóza, skolióza. Sklopená poloha. Plýtvání svalů. | Denervace myopatie, nepoužívejte atrofii |
| Kardiovaskulární systém | Zrychlený hypertenze. Dilatace kořene aorty. Kardiomyopatie. | Zvýšené intima mediální zahušťování. Ateroskleróza, arterioskleróza. |
| Gastrointestinální systém | Těžká reflux. Abnormální gastrointestinální motilita. Mnozí mají perkutánně gastrostomické trubičky. Hepatomegalie, splenomegalie, zvýšené jaterní enzymy. Změněno metabolismus drog | - |
| Renální systém | Selhání ledvin | Ledvinové tepny vykazují změny v pokročilé ateroskleróze a arterioskleróze. Jednostranný nebo hypoplastické ledviny. |
| Rozmnožovací systém | - | - |
| Muži | Micropenis, menší testikulární velikost | - |
| Ženy | Ovariální atrofie. Úspěšný těhotenství byl nahlášen. | - |
| Endokrinní systémy | Normální sekundární sexuální charakteristiky. Normální růstový hormon, hormon stimulující štítnou žlázu, hladiny vápníku | Normální hypofýza a štítná žláza |
| Ekrinní systémy | Snížení produkce potu, slzy, sliny | - |
Viz také
- Zrychlené stárnutí nemoci
- Biogerontologie
- Degenerativní onemocnění
- Genetická porucha
- CAMFAK syndrom - považována za formu (nebo podmnožinu) Cockaynova syndromu[20]
Reference
- ^ Bertola; Cao, H; Albano, Lm; Oliveira, Dp; Kok, F; Marques-Dias, Mj; Kim, Ca; Hegele, Ra (2006). „Cockayneův syndrom typu A: nové mutace u osmi typických pacientů“. Journal of Human Genetics. 51 (8): 701–5. doi:10.1007 / s10038-006-0011-7. PMID 16865293.
- ^ James, William; Berger, Timothy; Elston, Dirk (2005). Andrewsovy nemoci kůže: Klinická dermatologie (10. vydání). Saunders. p.575. ISBN 978-0-7216-2921-6.
- ^ A b C d E Bender M, Potocki L, Metry D. O jaký syndrom jde? Cockaynův syndrom. Dětská dermatologie [seriál online]. Listopad 2003; 20 (6): 538-540. Dostupné z: MEDLINE s plným textem, Ipswich, MA. Zpřístupněno 30. dubna 2015.
- ^ Hoeijmakers JH (říjen 2009). „Poškození DNA, stárnutí a rakovina“. N. Engl. J. Med. 361 (15): 1475–85. doi:10.1056 / NEJMra0804615. PMID 19812404.
- ^ A b C d Nance M, Berry S (1. ledna 1992). „Cockaynův syndrom: recenze 140 případů“. American Journal of Medical Genetics. 42 (1): 68–84. doi:10,1002 / ajmg.1320420115. PMID 1308368.
- ^ A b C d Neill CA, Dingwall MM. Syndrom připomínající progérii: přehled dvou případů. Archiv nemocí v dětství. 1950; 25 (123): 213-223.
- ^ A b C d E F Cockaynův syndrom. Genetická domácí reference http://ghr.nlm.nih.gov/condition/cockayne-syndrome Publikováno 28. dubna 2015. Recenzováno: 5. května 2010. Zpřístupněno 30. dubna 2015.
- ^ A b C d E F Syndrom Javadzadeh M. Cockayne. Írán J Dítě Neurol. Podzim 2014; 8; 4 (Suppl.1): 18-19.
- ^ Cerebrooculofacioskeletální syndrom 2. Mendelova dědičnost online u člověka. https://omim.org/entry/610756. Publikováno 2. 12. 2007.
- ^ Laugel V. Cockayne syndrom. 2000 28. prosince [Aktualizováno 2012 14. června]. In: Pagon RA, Adam MP, Ardinger HH a kol., Redaktoři. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. Dostupný z: [1]
- ^ van Hoffen A, Natarajan AT, Mayne LV, van Zeeland AA, Mullenders LH, Venema J (1993). „Nedostatečná oprava transkribovaného řetězce aktivních genů v buňkách Cockayneova syndromu“. Nucleic Acids Res. 21 (25): 5890–5. doi:10.1093 / nar / 21.25.5890. PMC 310470. PMID 8290349.
- ^ A b Iyama T, Wilson DM (2016). „Prvky, které regulují reakci DNA na poškození proteinů defektních u Cockaynova syndromu“. J. Mol. Biol. 428 (1): 62–78. doi:10.1016 / j.jmb.2015.11.020. PMC 4738086. PMID 26616585.
- ^ Iyama T, Lee SY, Berquist BR, Gileadi O, Bohr VA, Seidman MM, McHugh PJ, Wilson DM (2015). „CSB interaguje s SNM1A a podporuje zpracování křížového propojení DNA mezi vlákny“. Nucleic Acids Res. 43 (1): 247–58. doi:10.1093 / nar / gku1279. PMC 4288174. PMID 25505141.
- ^ Batenburg NL, Thompson EL, Hendrickson EA, Zhu XD (2015). „Protein skupiny B podle Cockayneova syndromu reguluje opravu dvouvláknového zlomu DNA a aktivaci kontrolního bodu“. EMBO J.. 34 (10): 1399–416. doi:10.15252 / embj.201490041. PMC 4491999. PMID 25820262.
- ^ Wei L, Nakajima S, Böhm S, Bernstein KA, Shen Z, Tsang M, Levine AS, Lan L (2015). „Poškození DNA během fáze G0 / G1 spouští homologní rekombinaci závislou na RNA, Cockayneův syndrom závislý na B“. Proc. Natl. Acad. Sci. USA. 112 (27): E3495–504. Bibcode:2015PNAS..112E3495W. doi:10.1073 / pnas.1507105112. PMC 4500203. PMID 26100862.
- ^ Kyllermen, Marten. Cockaynův syndrom. Švédské informační centrum pro vzácná onemocnění. 2012: 4,0. http://www.socialstyrelsen.se/rarediseases/cockaynesyndrome#anchor_17 Archivováno 24. 09. 2015 na Wayback Machine
- ^ Název: Cockayne Syndrome Autoři: Dr Nita R Sutay, Dr Md Ashfaque Tinmaswala, Dr Manjiri Karlekar, Dr Swati Jhahttp: //jmscr.igmpublication.org/v3-i7/35%20jmscr.pdf
- ^ "Cockaynův syndrom | Informační centrum o genetických a vzácných onemocněních (GARD) - program NCATS".
- ^ Karikkineth, A. C .; Scheibye-Knudsen, M .; Fivenson, E .; Croteau, D. L .; Bohr, V. A. (2016). „Cockayneův syndrom: Klinické rysy, modelové systémy a cesty“. Recenze výzkumu stárnutí. 33: 3–17. doi:10.1016 / j.arr.2016.08.002. PMC 5195851. PMID 27507608.
- ^ „Orphanet: CAMFAK syndrom“.
externí odkazy
- Tento článek včlení nějaký public domain text z Americká národní lékařská knihovna
| Klasifikace | |
|---|---|
| Externí zdroje |