Nedostatek translokázy ornitinu - Ornithine translocase deficiency
tento článek potřebuje další citace pro ověření. (Březen 2008) (Zjistěte, jak a kdy odstranit tuto zprávu šablony) |
| Nedostatek translokázy ornitinu | |
|---|---|
| Ostatní jména | Syndrom HHH, nedostatek ORNT1, nedostatek nosiče ornitinu, syndrom trojitého H. |
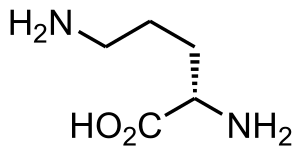 | |
| Ornitin | |
Nedostatek translokázy ornitinu, také zvaný hyperornithinemie-hyperamonemie-homocitrullinurie (HHH) syndrom,[1] je vzácný autosomální recesivní[2] porucha močovinového cyklu ovlivňující enzym ornitin translocase, Který způsobuje amoniak hromadit se v krvi, stav zvaný hyperamonémie.
Amoniak, který se tvoří při rozpadu bílkovin v těle, je toxický, pokud jsou jeho hladiny příliš vysoké. The nervový systém je obzvláště citlivý na účinky přebytečného amoniaku.
Patofyziologie

Mutace v SLC25A15 způsobit ornitin nedostatek translokázy.[3] Deficit translokázy ornithinu patří do třídy metabolické poruchy označované jako poruchy močovinového cyklu. Cyklus močoviny je sled reakcí, ke kterým dochází v játra buňky. Tento cyklus zpracovává přebytečný dusík, generovaný při použití bílkovin v těle, za vzniku sloučeniny zvané močovina, která je vylučována ledviny. The SLC25A15 Gen poskytuje pokyny pro výrobu proteinu nazývaného mitochondriální transportér ornitinu. Tento protein je potřebný k pohybu molekuly zvané ornitin uvnitř mitochondrie (centra produkce energie v buňkách). Konkrétně tento protein transportuje ornitin přes vnitřní membránu mitochondrií do oblasti zvané mitochondriální matice, kde se účastní močovinového cyklu. Mutace v SLC25A15 Výsledkem genu je mitochondriální transportér ornitinu, který je nestabilní nebo má špatný tvar a který nemůže přivést ornitin do mitochondriální matrice. Toto selhání transportu ornithinu způsobuje přerušení močovinového cyklu a hromadění amoniaku, což má za následek známky a příznaky deficitu translokázy ornithinu.[Citace je zapotřebí ]
Tato porucha se dědí v autosomálně recesivní vzor, což znamená, že defektní gen je umístěn na autosome a dvě kopie genu - jedna od každého rodiče - se musí narodit s poruchou. Rodiče jedince s autosomálně recesivní poruchou nesou vždy jednu kopii změněného genu, ale nevykazují známky a příznaky poruchy.
Diagnóza
Klinické nálezy u syndromu HHH jsou nespecifické. Pokud existuje podezření na poruchu, mohou laboratorní testy poskytnout diagnostické informace. Analýza aminokyselin v plazmě ukáže zvýšené hladiny ornitinu a aminokyseliny v moči detekují homocitrulin. Může být také zvýšena kyselina orotová. Hladiny amoniaku mohou být variabilně zvýšeny. Pokud jsou tyto nálezy k dispozici, mohou další potvrzující informace poskytnout molekulární testy.[Citace je zapotřebí ]
Léčba
Léčba zahrnuje přerušení příjmu bílkovin, intravenózní infuzi glukózy a podle potřeby infuzi doplňkového argininu a léků na odstranění amoniaku, fenylacetát sodný a benzoát sodný.
Viz také
- Nedostatek ornithin-transkarbamylázy
- Vrozené poruchy metabolismu
- Nedostatek ornitinaminotransferázy (gyrátová atrofie cévnatky a sítnice)
Reference
- ^ Online Mendelian Inheritance in Man (OMIM): 238970
- ^ Hommes FA, Roesel RA, Metoki K, Hartlage PL, Dyken PR (únor 1986). "Studie na případu HHH syndromu (hyperamonémie, hyperornithinémie, homocitrullinurie)". Neuropediatrie. 17 (1): 48–52. doi:10.1055 / s-2008-1052499. ISSN 0174-304X. PMID 3960284.
- ^ Smith, L. D .; Garg, U. (01.01.2017), Garg, Uttam; Smith, Laurie D. (eds.), „Kapitola 5 - Močovinový cyklus a další poruchy hyperamonémie“, Biomarkery u vrozených chyb metabolismu, San Diego: Elsevier, s. 103–123, doi:10.1016 / b978-0-12-802896-4.00004-3, ISBN 978-0-12-802896-4, vyvoláno 2020-11-10
- Charles Scriver Beaudet, A.L., Valle, D., Sly, W.S., Vogelstein, B., Childs, B., Kinzler, K.W. (přístup 2007). New York: McGraw-Hill. Souhrny 255 kapitol, plný text na mnoha univerzitách. K dispozici je také Blog OMMBID.
Další čtení
- Nedostatek translokázy ornitinu na NLM Genetická domácí reference
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |