Nedostatek ornitinaminotransferázy - Ornithine aminotransferase deficiency
| Nedostatek ornitinaminotransferázy | |
|---|---|
| Ostatní jména | Atrofie gyrátů (cévnatky a sítnice) |
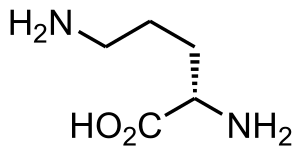 | |
| Ornitin | |
| Specialita | Oftalmologie, lékařská genetika |
Nedostatek ornitinaminotransferázy (také známý jako gyrátová atrofie cévnatky a sítnice) je vrozená chyba ornitin metabolismus způsobený sníženou aktivitou enzym ornitinaminotransferáza. Biochemicky jej lze detekovat zvýšenou hladinou ornithinu v krvi.[1] Klinicky se projevuje zpočátku špatně noční vidění, která pomalu postupuje k úplné slepotě.[2] Předpokládá se, že je zděděn v autosomálně recesivní způsob. V literatuře bylo popsáno přibližně 200 známých případů. Výskyt je nejvyšší v Finsko, odhaduje se na 1:50 000.[2]
Výzkum naznačuje, že mohou existovat určité nepříznivé účinky na svaly a také na mozek. Příčina je poněkud nejasná, ale může souviset s velmi nízkou úrovní kreatin často nalezené v této populaci.
Léčba může zahrnovat vitamin B6, lysin nebo dramatická změna ve stravě, aby se minimalizovala arginin ze stravy pacientů. Výzkum ukázal, že tato léčba může být poněkud účinná při snižování koncentrací ornitinu v krvi u některých pacientů, ať už v kombinaci nebo jednotlivě. Bylo zjištěno, že vitamin B6 je velmi účinný u malé části pacientů.
Prezentace
Poměrně často je přítomným příznakem nedostatku ornitinaminotransferázy (OAT) krátkozrakost který postupuje do Noční slepota. Nástup krátkozrakosti je často v raném dětství. Oftalmologické nálezy u postižených jedinců zahrnují zúžená zorná pole, zadní subkapsulární šedý zákal (může začít v pozdním mladistvém věku), zvýšené temné adaptační prahy a snížena nebo chybí elektroretinografické odpovědi.[3] Příznaky nedostatku OAT jsou progresivní a ve věku 45 až 65 let je většina postižených jedinců téměř úplně slepý.[3]
V některých případech se u postižených jedinců projeví v novorozeneckém období onemocnění, které velmi napodobuje klasický defekt močovinového cyklu, jako je nedostatek ornithin-transkarbamylázy, protože blokování metabolismu ornithinu vede k sekundární dysfunkci močovinový cyklus. Tito jedinci jsou přítomni hyperamonémie, špatné krmení, neprospívání a zvýšené vylučování kyselina orotová.[3]
Genetika
Nedostatek OAT se dědí v autosomálně recesivní způsobem, což znamená, že postižený jedinec musí zdědit mutovaného alela od obou rodičů. Enzym, ornitinaminotransferáza je kódován pro gen OVES, umístěný na 10q26. Nedostatek OAT má zvýšený výskyt v Finsko,[2] a tato populace má společné mutace což představuje více než 85% mutovaných alel v této populaci. U žádné jiné populace to nebylo popsáno.[3]
Diagnóza
Na základě klinického podezření bude diagnostické testování často sestávat z měření aminokyselina koncentrace v plazma, při hledání výrazně zvýšené ornitin koncentrace. Měření moč Koncentrace aminokyselin jsou někdy nutné, zejména v případech nástupu novorozence k identifikaci přítomnosti nebo nepřítomnosti homocitrulin za vyloučení nedostatek ornitinové translokázy (hyperornithinemie, hyperamonemie, syndrom homocitrullinurie, HHH syndrom).[3] Koncentrace ornithinu tedy mohou být nespolehlivým indikátorem v novorozeneckém období screening novorozenců nemusí tento stav detekovat, i když je v screeningovém panelu zahrnut ornithin. Enzymové testy pro měření aktivity ornitinaminotransferázy lze provádět z fibroblasty nebo lymfoblasty pro potvrzení nebo během novorozeneckého období, kdy jsou výsledky biochemických testů nejasné.[3] Možné je také molekulárně genetické testování.[3]
Léčba
Ke snížení hladiny ornithinu v krvi byla použita strava omezená na arginin.[2] Některé výzkumy ukázaly, že pokud je dieta nebo jiná léčba zahájena v raném věku, lze výsledek zlepšit.[2]
Reference
- ^ „Gyrátová atrofie cévnatky a sítnice“. Národní institut zdraví. Citováno 2012-08-23.
- ^ A b C d E „# 288870 - Gyrátová atrofie cévnatky a sítnice“. Univerzita Johna Hopkinse. Citováno 2012-08-23.
- ^ A b C d E F G Baumgartner, Matthias R .; Valle, David (2012). "Poruchy metabolismu ornitinu". V Saudubray, Jean-Marie; van den Berghe, Georges; Walter, John H. (eds.). Vrozené metabolické nemoci: diagnostika a léčba (5. vydání). New York: Springer. 323–332. ISBN 978-3-642-15719-6.
externí odkazy
| Klasifikace |
|---|