Noonanův syndrom s více lentiginy - Noonan syndrome with multiple lentigines
| Noonanův syndrom s více lentiginy (NSML) | |
|---|---|
| Ostatní jména | Syndrom LEOPARD, kardiokutánní syndrom, Gorlinův syndrom II, syndrom lentiginosis profusa, progresivní kardiomyopatická lentiginóza,[1]:550 Capute-Rimoin-Konigsmark-Esterly-Richardsonův syndrom, Moynahanův syndrom |
 | |
| Tříčtvrtinový pohled na obličej, pacient první generace vykazuje mírný obraz prognathismus a nízko nasazené uši | |
| Specialita | Lékařská genetika |
Noonanův syndrom s více lentiginy (NSML), která je součástí skupiny zvané Ras /MAPK syndromy dráhy,[2] je vzácný autosomálně dominantní,[3] multisystémové onemocnění způsobené a mutace v protein tyrosin fosfatáza, nereceptorový gen typu 11 (PTPN11 ). Toto onemocnění je komplex funkcí, většinou zahrnujících kožní, kosterní a kardiovaskulární systém, které se mohou nebo nemusí vyskytovat u všech pacientů. Povaha toho, jak mutace způsobuje každý z příznaků onemocnění, není dobře známa; výzkum však pokračuje. Je to RASopatie.
Noonanův syndrom s vícečetnými lentiginy je způsoben jiným missense mutace stejného genu. Noonanův syndrom je poměrně běžný (1: 1 000 až 1: 2 500 živě narozených dětí) a neurofibromatóza 1 (o kterém se kdysi myslelo, že souvisí s NSML) je také běžný (1: 3500); avšak ne epidemiologické existují data pro NSML.[4]
Příznaky a symptomy
Alternativní název stavu, syndrom LEOPARD, je a mnemotechnická pomůcka, původně vytvořen v roce 1969,[5] protože tento stav je charakterizován některými z následujících sedmi stavů, jejichž první písmena znamenají LEOPARD spolu s charakteristikou „piha "kůže způsobené lentiginy to připomíná velká kočka.
- Lentigines - Červenohnědý až tmavě hnědý makuly (povrchová kůže léze ) obvykle se vyskytující ve vysokém počtu (10 000+) na velké části pokožky, někdy vyšší než 80%. Mohou se dokonce objevit uvnitř úst (bukální ) nebo na povrchu oka (sklerální ). Ty mají nepravidelné okraje a velikost od 1 mm v průměru do café-au-lait skvrny, o průměru několika centimetrů. Také některé oblasti vitiligo -jako hypopigmentace lze pozorovat.
- Elektrokardiografické vedení abnormality: Obecně pozorováno na elektrokardiograf jako blok větve svazku.
- Oční hypertelorismus: Širokoúhlé oči, které vedou k podobné obličejové podobnosti mezi pacienty. Abnormality obličeje jsou druhým nejvýznamnějším příznakem po lentiginy. Abnormality také zahrnují: široký nosní kořen, prognathismus (vyčnívající dolní čelist), nebo nízko posazené, případně otočené, uši.
- Plicní stenóza: Zúžení plicní tepna jak opouští srdce. Mohou být přítomny další srdeční abnormality, včetně aortální stenóza nebo výhřez mitrální chlopně.
- Abnormální genitálie: obvykle kryptorchismus (zadržení varlata v těle) nebo monorchismus (jedno varle). U pacientek se to projevuje jako chybějící nebo jednotlivé vaječníky, které je přirozeně mnohem těžší detekovat. Ultrazvukové zobrazování se provádí v pravidelných intervalech od 1 roku, aby se zjistilo, zda jsou přítomny vaječníky.
- Zpomalený růst: Pomalý nebo zakrnělý růst. Většina novorozenců s tímto syndromem má normální porodní hmotnost a délku, ale během prvního roku se často zpomalí.
- Hluchota: Senzorineurální (nervová hluchota).
Přítomnost všech těchto charakteristické znaky není nutná pro diagnózu. Klinické diagnóza se považuje za vyrobenou, když s lentiginy v současnosti jsou pozorovány 2 další příznaky, jako jsou abnormality EKG a oční hypertelorismus, nebo bez lentiginů, jsou přítomny 3 z výše uvedených stavů, přičemž u příbuzného prvního stupně (tj. rodiče, dítěte, sourozence) je klinická diagnóza.[6]
- Další dermatologické abnormality (axilární pihy, lokalizované hypopigmentace, interdigitální popruh hyperelastická kůže)
- Mírná mentální retardace je pozorována asi u 30% pacientů postižených tímto syndromem
- Nystagmus (nedobrovolné pohyby očí), záchvaty nebo hyposmie (snížená čichová schopnost) byla dokumentována u několika pacientů
- V roce 2004 byl pacient hlášen s opakující se horní končetinou aneuryzma to vyžadovalo chirurgické opravy.[7]
- V roce 2006 byl hlášen pacient s NSML akutní myeloidní leukémie.[8]
Vzhledem ke vzácnosti samotného syndromu je těžké určit, zda jsou některá další onemocnění skutečně součástí syndromu. Se základní populací, která může mít méně než jeden tisíc jedinců, může jeden nebo dva odlehlé případy statistickou populaci velmi rychle vychýlit.

Zobrazení ruky 37letého pacienta interdigitální popruh

37letý pacient (druhá generace), vykazující hypertelorismus, široký kořen nosu, mírnou ptózu
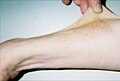
Třicet sedm let starý pacient předvádí hyperelasticity

21měsíční pacient třetí generace, potvrzeno genetickými testy jako Y279C, vykazující oční hyperteliorismus, cefalofaciální podobnost.

Trup třicet sedm let starý pacient druhé generace, vystavující lentiginóza.




Patofyziologie

U dvou převládajících mutací NSML (Y279C a T468M ) mutace způsobí ztrátu katalytická aktivita SHP2 proteinu (genový produkt z PTPN11 gen), což je dříve nerozpoznané chování pro tuto třídu mutací.[9] To narušuje růstový faktor a související signalizaci. Zatímco další výzkumy tento mechanismus potvrzují,[10][11] je zapotřebí další výzkum, aby se zjistilo, jak to souvisí se všemi pozorovanými účinky NSML.
Diagnóza
Přítomnost onemocnění lze potvrdit genetickým testem. Ve studii s 10 kojenci s klinickými indikacemi NSML před jejich prvními narozeninami bylo u 8 (80%) pacientů potvrzeno podezření na mutaci. Následně bylo zjištěno, že má další pacient s podezřením na mutaci NF1, po zhodnocení matky.[12]
Je jich identifikováno 5 alelický varianty odpovědný za NSML. Y279C, T468M, A461T, G464A, a Q510P což se jeví jako jedinečná familiární mutace v tom, že všechny ostatní varianty jsou spíše způsobeny chybami přechodu než transverze.
Léčba
Navrhuje se, aby po diagnostikování jednotlivci byli běžně sledováni jako příznaky kardiologem, endokrinologem, dermatologem a dalšími příslušnými specializacemi.
Doporučuje se, aby osoby se syndromem, které jsou schopné mít děti, vyhledaly genetické poradenství, než se rozhodnou mít děti. Vzhledem k tomu, že se syndrom často vyskytuje jako forma fruste (neúplná nebo neobvyklá forma) varianta, musí být provedeno vyšetření všech členů rodiny.[13] Jako autozomálně dominantní rys existuje u každého dítěte padesátiprocentní šance, že se také narodí se syndromem. I když je plně penetrantní, protože syndrom má proměnlivou expresivitu, jedna generace může mít mírnou expresi syndromu, zatímco další může být hluboce ovlivněna.
Jakmile je učiněno rozhodnutí mít děti a pár počne, je během těhotenství sledován plod na vyšetření srdce. Pokud se zjistí závažná srdeční malformace, dostanou rodiče radu ohledně pokračování v těhotenství.
Jiná léčba je rutinní péče, protože jsou přítomny příznaky:[13]
- Pro osoby s endokrinními problémy (nízká hladina thyrotopin [hormon hypofýzy odpovědný za regulaci hormonů štítné žlázy], hormon stimulující folikuly ) se doporučuje léková terapie.
- Pro ty, kteří jsou narušeni výskytem lentiginů, může být prospěšná kryochirurgie. Vzhledem k velkému počtu lentiginů to může být časově náročné. Může pomoci alternativní léčba krémy tretinoinem nebo hydrochinonem.
- Lékové terapie u pacientů se srdečními abnormalitami, protože tyto abnormality jsou natolik závažné, že vyžadují použití těchto terapií. EKG jsou zpravidla před chirurgickým zákrokem povinné arytmie.
Prognóza
Samotný NSML není život ohrožující diagnóza, většina lidí s diagnostikovaným stavem žije normální život. Obstrukční kardiomyopatie a další patologické nálezy postihující kardiovaskulární systém mohou být příčinou smrti u těch, jejichž srdeční deformity jsou hluboké.[13]
Epidemiologie
Různá literatura popisuje syndrom jako „vzácný“[13] nebo „extrémně vzácné“.[14] Nejsou k dispozici žádné epidemiologické údaje o tom, kolik lidí trpí syndromem na celém světě; v lékařské literatuře je však popsáno přibližně 200 případů.[15]
Dějiny
Zeisler a Becker nejprve popsali syndrom s vícečetným syndromem lentiginy, hypertelorismus, pectus carinatum (vyčnívající hrudní kost) a prognathismus (výčnělek dolní čelisti) v roce 1936.[16] V průběhu let byly přidávány sporadické popisy. V roce 1962 byly se stavem poprvé spojeny srdeční abnormality a nízká postava.[17] V roce 1966 byly přidány tři rodinné případy, matka, její syn a dcera.[18] V roce 1968 byl přidán další případ matky ke dvěma odděleným dětem s různým otcovstvím obou dětí.[19]
Věřilo se až v roce 2002[20] že Noonanův syndrom s více lentiginy (NSML) souvisel neurofibromatóza typu I (von Recklinghausenův syndrom). Ve skutečnosti, protože obojí ICD9 a ICD10 chybí specifický diagnostický kód pro NSML, diagnostický kód pro NF1 se stále někdy používá pro diagnostické účely, i když se ukázalo, že gen není spojen s NF1 místo.[21]
Viz také
Reference
- ^ James, William; Berger, Timothy; Elston, Dirk (2005). Andrewsovy choroby kůže: Klinická dermatologie (10. vydání). Saunders. ISBN 0-7216-2921-0.
- ^ Tidyman WE, Rauen KA (červen 2009). „RASopathies: vývojové syndromy dysregulace dráhy Ras / MAPK“. Aktuální názor na genetiku a vývoj. 19 (3): 230–6. doi:10.1016 / j.gde.2009.04.001. PMC 2743116. PMID 19467855.
- ^ Coppin BD, Temple IK (1997). „Mnohočetný lentiginový syndrom (syndrom LEOPARD nebo progresivní kardiomyopatická lentiginóza)“. Journal of Medical Genetics. 34 (7): 582–6. doi:10,1136 / jmg. 34,7,582. PMC 1051000. PMID 9222968.
- ^ Tullu MS, Muranjan MN, Kantharia VC a kol. (1. dubna 2000). „Neurofibromatóza-Noonanův syndrom nebo LEOPARDOVÝ syndrom? Klinické dilema“. J Postgrad Med. 46 (2): 98–100. PMID 11013475.
- ^ Gorlin RJ, Anderson RC, Blaw M (1969). "Syndrom více lentigenů". Dopoledne. J. Dis. Dítě. 117 (6): 652–62. doi:10.1001 / archpedi.1969.02100030654006. PMID 5771505.
- ^ Voron DA, Hatfield HH, Kalkhoff RK (1976). "Syndrom více lentiginů. Kazuistika a přehled literatury". Dopoledne. J. Med. 60 (3): 447–56. doi:10.1016/0002-9343(76)90764-6. PMID 1258892.
- ^ Yagubyan M, Panneton JM, Lindor NM, Conti E, Sarkozy A, Pizzuti A (duben 2004). „LEOPARD syndrom: nová asociace polyaneuryzmatu a aktualizace molekulární genetiky onemocnění“. J. Vasc. Surg. 39 (4): 897–900. doi:10.1016 / j.jvs.2003.11.030. PMID 15071461.
- ^ Uçar C, Calýskan U, Martini S, Heinritz W (březen 2006). „Akutní myelomonocytová leukémie u chlapce se syndromem LEOPARD (pozitivní mutace genu PTPN11)“. J. Pediatr. Hematol. Oncol. 28 (3): 123–5. doi:10.1097 / 01.mph.0000199590.21797.0b. PMID 16679933. S2CID 21559684.
- ^ Tartaglia M, Martinelli S, Stella L a kol. (2006). „Rozmanitost a funkční důsledky zárodečných a somatických mutací PTPN11 u lidských onemocnění“. American Journal of Human Genetics. 78 (2): 279–90. doi:10.1086/499925. PMC 1380235. PMID 16358218.
- ^ Hanna N, Montagner A, Lee WH a kol. (2006). "Snížená aktivita fosfatázy SHP-2 u syndromu LEOPARD: důsledky pro vazbu PI3K na Gab1". FEBS Lett. 580 (10): 2477–82. doi:10.1016 / j.febslet.2006.03.088. PMID 16638574. S2CID 27676871.
- ^ Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG (2006). „PTPN11 (Shp2) mutace v syndromu LEOPARD mají dominantní negativní, neaktivační účinky“. J. Biol. Chem. 281 (10): 6785–92. doi:10,1074 / jbc.M513068200. PMID 16377799.
- ^ Digilio MC, Sarkozy A, de Zorzi A a kol. (2006). „LEOPARD syndrom: klinická diagnóza v prvním roce života“. American Journal of Medical Genetics. 140 (7): 740–6. doi:10,1002 / ajmg.a.31156. PMID 16523510. S2CID 19570040.
- ^ A b C d LEOPARDOVÝ syndrom na eMedicína
- ^ "LEOPARDOVÝ syndrom". NORD - Národní organizace pro vzácné poruchy.
- ^ „Noonanův syndrom s více lentiginy“. Americká národní lékařská knihovna.
- ^ Zeisler EP, Becker SW (1936). "Generalizované lentigo: jeho vztah k systémovým nevyvýšeným névům". Arch Dermatol Syphilol. 33: 109–125. doi:10.1001 / archderm.1936.01470070112010.
- ^ Moynahan EJ (1962). „Několik symetrických krtků s psychickým a somatickým infantilismem a hypoplázií genitálií: první mužský případ nového syndromu“. Sborník Královské lékařské společnosti. 55 (11): 959–960. doi:10.1177/003591576205501112. PMC 1896920. PMID 19994192.
- ^ Walther RJ, Polansky BJ, Grotis IA (1966). "Elektrokardiografické abnormality v rodině s generalizovaným lentigem". N. Engl. J. Med. 275 (22): 1220–5. doi:10.1056 / NEJM196612012752203. PMID 5921856.
- ^ Matthews NL (1968). "Lentigo a elektrokardiografické změny". N. Engl. J. Med. 278 (14): 780–1. doi:10.1056 / NEJM196804042781410. PMID 5638719.
- ^ National Library of Medicine MeSH: C05.660.207.525
- ^ Ahlbom BE, Dahl N, Zetterqvist P, Annerén G (1995). „Noonanův syndrom se skvrnami v kavárně-au-lait a syndromem více lentiginů není spojen s lokusem neurofibromatózy typu 1“. Clin. Genet. 48 (2): 85–9. doi:10.1111 / j.1399-0004.1995.tb04061.x. PMID 7586657. S2CID 31291484.
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |
- NSML na NIH /UW Genové testy
- Gorlinův syndrom II na Kdo to pojmenoval?
- DermAtlas 981603547
- Dermnetnz
- DermIS