MT-ATP8 - MT-ATP8 - Wikipedia

| ATP syntázový protein 8 (metazoa) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifikátory | |||||||||
| Symbol | ATP-synt_8 | ||||||||
| Pfam | PF00895 | ||||||||
| Pfam klan | CL0255 | ||||||||
| InterPro | IPR001421 | ||||||||
| |||||||||
| Rostlina ATP syntáza F0 podjednotka 8 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifikátory | |||||||||
| Symbol | YMF19 | ||||||||
| Pfam | PF02326 | ||||||||
| Pfam klan | CL0255 | ||||||||
| InterPro | IPR003319 | ||||||||
| |||||||||
| Plísňový protein ATP syntázy 8 (A6L) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifikátory | |||||||||
| Symbol | Fun_ATP-synt_8 | ||||||||
| Pfam | PF05933 | ||||||||
| Pfam klan | CL0255 | ||||||||
| InterPro | IPR009230 | ||||||||
| |||||||||
MT-ATP8 (nebo ATP8) je mitochondriální gen s celým názvem „mitochondriálně kódovaná membránová podjednotka ATP syntázy 8“, která kóduje podjednotku mitochondriální ATP syntáza, ATP syntáza FÓ podjednotka 8 (nebo podjednotka A6L). Tato podjednotka patří k FÓ komplex velkého transmembránového typu F. ATP syntáza.[2] Tento enzym, který je také známý jako komplex V, je zodpovědný za poslední krok oxidační fosforylace v elektronový transportní řetězec. Konkrétně jeden segment ATP syntázy umožňuje kladné nabití ionty, volala protony, protékat specializovanou membránou uvnitř mitochondrií. Další segment enzymu využívá energii vytvořenou tímto tokem protonů k přeměně molekuly zvané adenosindifosfát (ADP) do ATP.[3] Podjednotka 8 se liší v sekvence mezi Metazoa, rostliny a Houby.
Struktura
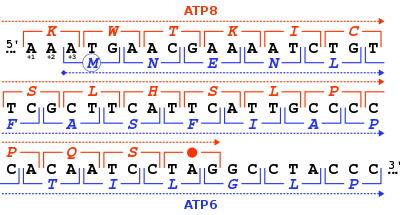
ATP syntázový protein 8 lidských a jiných savců je kódován v mitochondriální genom podle MT-ATP8 gen. Když byl poprvé publikován kompletní lidský mitochondriální genom, MT-ATP8 gen byl popsán jako neidentifikovaný čtecí rámec URF A6L.[2] Neobvyklá vlastnost MT-ATP8 gen se překrývá s 46 nukleotidy s MT-ATP6 gen. S ohledem na čtecí rámec (+1) z MT-ATP8, MT-ATP6 gen začíná na čtecím rámci +3.
Protein MT-ATP8 váží 8 kDa a skládá se z 68 aminokyseliny.[4][5] Protein je podjednotkou F1FÓ ATPáza, také známá jako Komplex V, který se skládá ze 14 jaderných a 2 mitochondriálně kódovaných podjednotek. ATPázy typu F se skládají ze dvou strukturálních domén, F1 obsahující extramembránové katalytické jádro a FÓ obsahující protonový kanál membrány, vzájemně spojené centrální stopkou a periferní stopkou. Jako podjednotka A je MT-ATP8 obsažen v nekatalytickém, transmembránový FÓ část komplexu zahrnující protonový kanál. Katalytická část mitochondriální ATP syntázy se skládá z 5 různých podjednotek (alfa, beta, gama, delta a epsilon) sestavených se stechiometrií 3 alfa, 3 beta a jediným zástupcem ostatních 3. Protonový kanál se skládá ze tří hlavní podjednotky (a, b, c). Tento gen kóduje delta podjednotku katalytického jádra. Alternativně byly identifikovány sestřihové varianty transkriptu kódující stejnou izoformu.[6][3]
Funkce
MT-ATP8 gen kóduje podjednotku mitochondrií ATP syntáza, který se nachází v tylakoidní membrána a vnitřní mitochondriální membrána. Mitochondriální ATP syntáza katalyzuje ATP syntéza, s využitím elektrochemický gradient z protony přes vnitřní membránu během oxidační fosforylace.[6] FÓ oblast způsobí rotaci F1, který má ve vodě rozpustnou složku, která hydrolyzuje ATP a společně F1FÓ vytváří dráhu pro pohyb protonů přes membránu.[7]
Tento protein podjednotka se jeví jako nedílná součást statorové stopky droždí mitochondriální F-ATPázy.[8] Stator statoru je ukotven v membrána, a působí tak, aby zabránil marné rotaci podjednotek ATPázy vzhledem k rotoru během syntézy / hydrolýzy ATP. Tato podjednotka může mít obdobnou funkci v Metazoa.
Nomenklatura
The nomenklatura enzymu má dlouhou historii. F1 zlomek odvozuje svůj název od výrazu „Frakce 1“ a FÓ (psaný jako dolní index „o“, nikoli „nula“) odvozuje svůj název od toho, že je vazbou zlomku pro oligomycin, typ přirozeně odvozeného antibiotika, které je schopné inhibovat FÓ jednotka ATP syntázy.[9][10] FÓ oblast ATP syntázy je protonový pór, který je zality v mitochondriální membráně. Skládá se ze tří hlavních podjednotek A, B a C a (u lidí) šesti dalších podjednotek, d, E, F, G, MT-ATP6 (nebo F6) a MT-ATP8 (nebo A6L). 3D struktura E-coli homolog této podjednotky byl modelován na základě elektronová mikroskopie data (řetězec M z PDB: 1c17). Tvoří transmembránový 4-α-svazek.
Klinický význam
Mutace na MT-ATP8 a další geny ovlivňující oxidační fosforylace v mitochondriích byly spojeny s řadou neurodegenerativní a kardiovaskulární poruchy, včetně deficitu mitochondriálního komplexu V, Leberova dědičná optická neuropatie (LHON), mitochondriální encefalomyopatie s cévními mozkovými příhodami (MELAS ), Leighův syndrom, a Syndrom NARP. Většina buněk těla obsahuje tisíce mitochondrií, každá s jednou nebo více kopiemi mitochondriální DNA. Závažnost některých mitochondriální poruchy je spojena s procentem mitochondrií v každé buňce, která má konkrétní genetickou změnu. Lidé s Leighův syndrom v důsledku mutace genu MT-ATP6 mívají velmi vysoké procento mitochondrií s mutací (od více než 90 procent do 95 procent). Méně závažné rysy NARP výsledkem nižšího procenta mitochondrií s mutací, obvykle 70 procent až 90 procent. Protože tyto dva stavy vyplývají ze stejných genetických změn a mohou se vyskytovat u různých členů jedné rodiny, vědci se domnívají, že mohou představovat spektrum překrývajících se rysů místo dvou odlišných syndromů.[3]
Nedostatek mitochondriálního komplexu V má heterogenní klinické projevy včetně neuropatie, ataxie, Hypertrofické kardiomyopatie. Hypertrofická kardiomyopatie se může projevovat zanedbatelně až extrémně hypertrofie, minimální až rozsáhlé fibróza a myocyt nepořádek, nepřítomnost až závažná obstrukce výtokového traktu levé komory a výrazné kontury / morfologie septa s extrémně odlišným klinickým průběhem.[11][12]
Nedostatek mitochondriálního komplexu V je nedostatek (nedostatek) nebo ztráta funkce u komplex V z elektronový transportní řetězec to může způsobit širokou škálu Příznaky a symptomy ovlivňující mnoho orgánů a systémů těla, zejména nervový systém a srdce. Porucha může být život ohrožující v kojeneckém věku nebo v raném dětství. Postižení jedinci mohou mít problémy s krmením, pomalý růst, nízký svalový tonus (hypotonie ), extrémní únava (letargie ), a vývojové zpoždění. Mají tendenci vyvíjet zvýšené hladiny kyselina mléčná v krvi (laktátová acidóza ), které mohou způsobit nevolnost, zvracení, slabost a rychlé dýchání. Vysoká úroveň amoniak v krvi (hyperamonémie ) se může také objevit u postižených jedinců a v některých případech může vést k abnormální funkci mozku (encefalopatie ) a poškození dalších orgánů.[13] Ataxie, mikrocefalie, u pacientů s mutací posunu snímků v MT-ATP6 bylo pozorováno zpoždění vývoje a mentální postižení. To způsobí vložení C na pozici 8612, což má za následek zkrácený protein dlouhý pouze 36 aminokyselin a dva T> C jedno-nukleotidové polymorfismy na pozicích 8610 a 8614, které vedou k homopolymerii cytosin protáhnout se.[14]
Hypertrofické kardiomyopatie, společný rys deficitu mitochondriálního komplexu V, je charakterizován zesílením (hypertrofie ) z srdeční sval to může vést k srdeční selhání.[13] Mutace m.8528T> C se vyskytuje v překrývající se oblasti genů MT-ATP6 a MT-ATP8 a byla popsána u více pacientů s infantilní kardiomyopatií. Tato mutace mění iniciační kodon v MT-ATP6 na threonin stejně jako změna z tryptofan na arginin na pozici 55 MT-ATP8.[15][12] Jedinci s nedostatkem mitochondriálního komplexu V mohou mít také charakteristický vzorec rysů obličeje, včetně vysokého čela, zakřiveného obočí, vnějších koutků očí směřujících dolů (downslanting oční štěrbiny ), výrazný hřbet nosu, nízko posazené uši, tenké rty a malá brada (micrognathia ).[13]
Infantilní hypertrofická kardiomyopatie (CMHI) je také způsobena mutacemi ovlivňujícími odlišné genetické loci, počítaje v to MT-ATP6 a MT-ATP8. Infantilní forma Hypertrofické kardiomyopatie, srdeční porucha charakterizovaná hypertrofie komor, který je obvykle asymetrický a často zahrnuje interventrikulární septum. Mezi příznaky patří dušnost, synkopa, kolaps, bušení srdce, a bolest na hrudi. Lze je snadno vyprovokovat cvičením. Porucha má inter- a intrafamiliální variabilitu od benigních po maligní formy s vysokým rizikem srdeční selhání a náhlá srdeční smrt.[11][12]
Reference
- ^ „Human PubMed Reference:“. Národní centrum pro biotechnologické informace, Americká národní lékařská knihovna.
- ^ A b Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (duben 1981). "Pořadí a organizace lidského mitochondriálního genomu". Příroda. 290 (5806): 457–65. Bibcode:1981 Natur.290..457A. doi:10.1038 / 290457a0. PMID 7219534. S2CID 4355527.
- ^ A b C „MT-ATP8“. Genetická domácí reference. NCBI.
- ^ Zong NC, Li H, Li H, Lam MP, Jimenez RC, Kim CS, Deng N, Kim AK, Choi JH, Zelaya I, Liem D, Meyer D, Odeberg J, Fang C, Lu HJ, Xu T, Weiss J , Duan H, Uhlen M, Yates JR, Apweiler R, Ge J, Hermjakob H, Ping P (říjen 2013). „Integrace biologie a medicíny srdečních proteomů pomocí specializované znalostní databáze“. Výzkum oběhu. 113 (9): 1043–53. doi:10.1161 / CIRCRESAHA.113.301151. PMC 4076475. PMID 23965338.
- ^ „ATP syntázový protein 8“. Srdeční organelární proteinová atlasová znalostní databáze (COPaKB).
- ^ A b "MT-ATP8 mitochondriálně kódovaná ATP syntáza 8 [Homo sapiens (člověk)]" ". Gen. NCBI.
- ^ Velours J, Paumard P, Soubannier V, Spannagel C, Vaillier J, Arselin G, Graves PV (květen 2000). „Organizace kvasinkové ATP syntázy F (0): studie založená na cysteinových mutantech, thiolové modifikaci a síťovacích činidlech“. Biochimica et Biophysica Acta (BBA) - bioenergetika. 1458 (2–3): 443–56. doi:10.1016 / S0005-2728 (00) 00093-1. PMID 10838057.
- ^ Stephens AN, Khan MA, Roucou X, Nagley P, Devenish RJ (květen 2003). „Molekulární sousedství podjednotky 8 kvasinkové mitochondriální F1F0-ATP syntázy sondované cysteinovou skenovací mutagenezí a chemickou modifikací“. The Journal of Biological Chemistry. 278 (20): 17867–75. doi:10,1074 / jbc.M300967200. PMID 12626501.
- ^ Kagawa Y, Racker E (květen 1966). "Částečné rozlišení enzymů katalyzujících oxidativní fosforylaci. 8. Vlastnosti faktoru, který uděluje citlivost na oligomycin mitochondriální adenosintrifosfatáze". The Journal of Biological Chemistry. 241 (10): 2461–6. PMID 4223640.
- ^ Mccarty RE (listopad 1992). „POHLED ROSTLINNÉHO BIOCHEMISTA NA H + -ATPázy A ATP SYNTÁZY“. The Journal of Experimental Biology. 172 (Pt 1): 431–441. PMID 9874753.
- ^ A b „MT-ATP8 - ATP syntázový protein 8 - Homo sapiens (člověk)“. www.uniprot.org. UniProt. Citováno 3. srpna 2018.
 Tento článek včlení text dostupný pod CC BY 4.0 licence.
Tento článek včlení text dostupný pod CC BY 4.0 licence. - ^ A b C Ware SM, El-Hassan N, Kahler SG, Zhang Q, Ma YW, Miller E, Wong B, Spicer RL, Craigen WJ, Kozel BA, Grange DK, Wong LJ (květen 2009). „Infantilní kardiomyopatie způsobená mutací v překrývající se oblasti mitochondriálních genů ATPázy 6 a 8“. Journal of Medical Genetics. 46 (5): 308–14. doi:10.1136 / jmg.2008.063149. PMID 19188198. S2CID 25354118.
- ^ A b C „Nedostatek mitochondriálního komplexu V“. Genetická domácí reference. NCBI. Citováno 3. srpna 2018.
 Tento článek včlení text z tohoto zdroje, který je v veřejná doména.
Tento článek včlení text z tohoto zdroje, který je v veřejná doména. - ^ Jackson CB, Hahn D, Schröter B, Richter U, Battersby BJ, Schmitt-Mechelke T, Marttinen P, Nuoffer JM, Schaller A (červen 2017). „Nová mitochondriální mutace posunu rámců ATP6 způsobující izolovaný nedostatek komplexu V, ataxii a encefalomyopatii“. European Journal of Medical Genetics. 60 (6): 345–351. doi:10.1016 / j.ejmg.2017.04.006. hdl:10138/237062. PMID 28412374.
- ^ Imai A, Fujita S, Kishita Y, Kohda M, Tokuzawa Y, Hirata T, Mizuno Y, Harashima H, Nakaya A, Sakata Y, Takeda A, Mori M, Murayama K, Ohtake A, Okazaki Y (březen 2016). „Rychle progresivní infantilní kardiomyopatie s nedostatkem V mitochondriálního dýchacího řetězce v důsledku ztráty proteinu ATPázy 6 a 8“. International Journal of Cardiology. 207: 203–5. doi:10.1016 / j.ijcard.2016.01.026. PMID 26803244.
Další čtení
- Torroni A, Achilli A, Macaulay V, Richards M, Bandelt HJ (červen 2006). "Sklizeň plodů lidského stromu mtDNA". Trendy v genetice. 22 (6): 339–45. doi:10.1016 / j.tig.2006.04.001. PMID 16678300.
- Bodenteich A, Mitchell LG, Polymeropoulos MH, Merril CR (květen 1992). „Dinukleotidová repetice v lidské mitochondriální D-smyčce“. Lidská molekulární genetika. 1 (2): 140. doi:10,1093 / hmg / 1,2,140-a. PMID 1301157.
- Lu X, Walker T, MacManus JP, Seligy VL (červenec 1992). „Diferenciace lidských buněk HT-29 adenokarcinomu tlustého střeva koreluje se zvýšenou expresí mitochondriální RNA: účinky trehalózy na růst a zrání buněk“. Výzkum rakoviny. 52 (13): 3718–25. PMID 1377597.
- Marzuki S, Noer AS, Lertrit P, Thyagarajan D, Kapsa R, Utthanaphol P, Byrne E (prosinec 1991). „Normální varianty lidské mitochondriální DNA a translačních produktů: budování referenční databáze“. Genetika člověka. 88 (2): 139–45. doi:10.1007 / bf00206061. PMID 1757091. S2CID 28048453.
- Moraes CT, Andreetta F, Bonilla E, Shanske S, DiMauro S, Schon EA (březen 1991). „Replikačně kompetentní lidská mitochondriální DNA bez oblasti promotoru těžkých řetězců“. Molekulární a buněčná biologie. 11 (3): 1631–7. doi:10.1128 / MCB.11.3.1631. PMC 369459. PMID 1996112.
- Attardi G, Chomyn A, Doolittle RF, Mariottini P, Ragan CI (1987). "Sedm neidentifikovaných čtecích rámců lidské mitochondriální DNA kóduje podjednotky dýchacího řetězce NADH dehydrogenázy". Cold Spring Harbor Symposia o kvantitativní biologii. 51 Pt 1 (1): 103–14. doi:10,1101 / sqb.1986.051.01.013. PMID 3472707.
- Chomyn A, Cleeter MW, Ragan CI, Riley M, Doolittle RF, Attardi G (říjen 1986). "URF6, poslední neidentifikovaný čtecí rámec lidské mtDNA, kóduje podjednotku NADH dehydrogenázy". Věda. 234 (4776): 614–8. Bibcode:1986Sci ... 234..614C. doi:10.1126 / science.3764430. PMID 3764430.
- Chomyn A, Mariottini P, Cleeter MW, Ragan CI, Matsuno-Yagi A, Hatefi Y, Doolittle RF, Attardi G (1985). „Šest neidentifikovaných čtecích rámců lidské mitochondriální DNA kóduje složky NADH dehydrogenázy dýchacího řetězce“. Příroda. 314 (6012): 592–7. Bibcode:1985 Natur.314..592C. doi:10.1038 / 314592a0. PMID 3921850. S2CID 32964006.
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (duben 1981). "Pořadí a organizace lidského mitochondriálního genomu". Příroda. 290 (5806): 457–65. Bibcode:1981 Natur.290..457A. doi:10.1038 / 290457a0. PMID 7219534. S2CID 4355527.
- Montoya J, Ojala D, Attardi G (duben 1981). "Charakteristické rysy 5'-koncových sekvencí lidských mitochondriálních mRNA". Příroda. 290 (5806): 465–70. Bibcode:1981 Natur.290..465M. doi:10.1038 / 290465a0. PMID 7219535. S2CID 4358928.
- Horai S, Hayasaka K, Kondo R, Tsugane K, Takahata N (leden 1995). „Nedávný africký původ moderních lidí odhalen úplnými sekvencemi hominoidních mitochondriálních DNA“. Sborník Národní akademie věd Spojených států amerických. 92 (2): 532–6. Bibcode:1995PNAS ... 92..532H. doi:10.1073 / pnas.92.2.532. PMC 42775. PMID 7530363.
- Rieder MJ, Taylor SL, Tobe VO, Nickerson DA (únor 1998). „Automatizace identifikace variací DNA pomocí kvalitního resekvencování fluorescence: analýza lidského mitochondriálního genomu“. Výzkum nukleových kyselin. 26 (4): 967–73. doi:10.1093 / nar / 26.4.967. PMC 147367. PMID 9461455.
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N (říjen 1999). "Reanalysis a revize cambridgeské referenční sekvence pro lidskou mitochondriální DNA". Genetika přírody. 23 (2): 147. doi:10.1038/13779. PMID 10508508. S2CID 32212178.
- Ingman M, Kaessmann H, Pääbo S, Gyllensten U (prosinec 2000). „Variace mitochondriálního genomu a původ moderních lidí“. Příroda. 408 (6813): 708–13. Bibcode:2000Natur.408..708I. doi:10.1038/35047064. PMID 11130070. S2CID 52850476.
- Finnilä S, Lehtonen MS, Majamaa K (červen 2001). „Fylogenetická síť pro evropskou mtDNA“. American Journal of Human Genetics. 68 (6): 1475–84. doi:10.1086/320591. PMC 1226134. PMID 11349229.
- Maca-Meyer N, González AM, Larruga JM, Flores C, Cabrera VM (2003). „Hlavní genomové mitochondriální linie určují časné lidské expanze“. Genetika BMC. 2: 13. doi:10.1186/1471-2156-2-13. PMC 55343. PMID 11553319.
- Herrnstadt C, Elson JL, Fahy E, Preston G, Turnbull DM, Anderson C, Ghosh SS, Olefsky JM, Beal MF, Davis RE, Howell N (květen 2002). „Reduced-median-network analysis of complete mitochondrial DNA coding-region addresses for the major African, Asian, and European haplogroups". American Journal of Human Genetics. 70 (5): 1152–71. doi:10.1086/339933. PMC 447592. PMID 11938495.
- Silva WA, Bonatto SL, Holanda AJ, Ribeiro-Dos-Santos AK, Paixão BM, Goldman GH, Abe-Sandes K, Rodriguez-Delfin L, Barbosa M, Paçó-Larson ML, Petzl-Erler ML, Valente V, Santos SE , Zago MA (červenec 2002). „Mitochondriální genomová rozmanitost domorodých Američanů podporuje jediný brzký vstup zakladatelských populací do Ameriky“. American Journal of Human Genetics. 71 (1): 187–92. doi:10.1086/341358. PMC 384978. PMID 12022039.
- Mishmar D, Ruiz-Pesini E, Golik P, Macaulay V, Clark AG, Hosseini S, Brandon M, Easley K, Chen E, Brown MD, Sukernik RI, Olckers A, Wallace DC (leden 2003). „Regionální variace mtDNA u lidí ve tvaru přirozeného výběru“. Sborník Národní akademie věd Spojených států amerických. 100 (1): 171–6. Bibcode:2003PNAS..100..171M. doi:10.1073 / pnas.0136972100. PMC 140917. PMID 12509511.
- Ingman M, Gyllensten U (červenec 2003). „Variace mitochondriálního genomu a evoluční historie australských a novoguinejských domorodců“. Výzkum genomu. 13 (7): 1600–6. doi:10,1101 / gr. 686603. PMC 403733. PMID 12840039.
Tento článek včlení text z United States National Library of Medicine, který je v veřejná doména.