Alfa-talasémie - Alpha-thalassemia
| Alfa-talasémie | |
|---|---|
| Ostatní jména | α-talasémie |
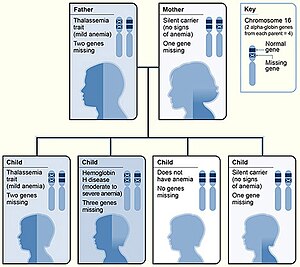 | |
| Vzorec dědičnosti alfa-talasémie | |
| Specialita | Hematologie |
| Příznaky | Žloutenka, únava[1] |
| Příčiny | Delece chromozomu 16p.[2] |
| Diagnostická metoda | Elektroforéza hemoglobinu[3] |
| Léčba | Krevní transfuze, možná splenektomie[1][4] |
Alfa-talasémie (α-talasémie, α-talasémie) je forma talasémie zahrnující geny HBA1[5] a HBA2.[6] Thalassemias jsou skupina zdědil krevní podmínky které mají za následek zhoršenou produkci hemoglobin, molekula, která nese kyslík v krvi.[7] Normální hemoglobin se skládá ze dvou alfa řetězce a dva beta řetězce; u alfa-talasémie existuje kvantitativní pokles množství alfa řetězců, což má za následek méně normálních molekul hemoglobinu. Alfa-talasémie navíc vede k produkci nestabilních molekul beta globinu, které způsobují zvýšenou hladinu červená krvinka zničení. Míra poškození závisí na tom, které klinické fenotyp je přítomen (kolik genů je ovlivněno).[3]
Příznaky a symptomy
Prezentace jedinců s alfa-talasemií se skládá z:
| Běžný | Neobvyklé |
|---|---|
|
|
Způsobit
Alfa-thalassemias se nejčastěji dědí v a Mendelian recesivní způsob. Jsou také spojeny s vypuštěním chromozom 16p.[2] Alfa thalassemii lze získat i za výjimečných okolností.[12]
Patofyziologie
Mechanismus vidí, že α thalassemias vede ke snížené produkci alfa-globinu, proto se produkuje méně řetězců alfa-globinu, což má za následek přebytek β řetězců u dospělých a přebytek γ řetězců u novorozenců. Přebytek β řetězců tvoří nestabilní tzv. Tetramery hemoglobin H nebo HbH ze čtyř beta řetězce. Přebytek y řetězců tvoří tetramery, které jsou špatnými nosiči O2 protože jejich afinita k O2 je příliš vysoká, takže není disociována na periferii. Homozygot α0 thalassemie, kde je mnoho γ4 ale vůbec se nevyskytují žádné a-globiny (označované jako HB Barts ), často končí smrtí krátce po narození.[3][1][13]
Diagnóza
Diagnóza alfa-talasémie je primárně laboratorním hodnocením a molekulární diagnostika. Alfa-thalassemii lze zaměnit anémie z nedostatku železa na úplném krevním obrazu nebo krevním filmu, protože oba stavy mají mikrocytární anémii. Sérové železo a sérový feritin lze použít k vyloučení anémie s nedostatkem železa.[3]
Typy
Existují dva genetické lokusy pro a globin, tedy čtyři alely jsou v diploidních buňkách. Dvě alely jsou mateřské a dvě alely jsou otcovského původu. Závažnost α-thalassemií koreluje s počtem postižených alel α-globinu: čím větší, tím závažnější budou projevy onemocnění. Když si všimneme genotypu, „α“ označuje funkční alfa řetězec a „-“ patologický.[1][13]
| Ovlivněny alely | Popis | Genotyp | |||||
|---|---|---|---|---|---|---|---|
| Jeden | Toto je známé jako alfa thalassemia tichý a u tohoto typu je účinek na syntézu hemoglobinu minimální. Tři geny pro a-globin jsou dostatečné pro umožnění normální produkce hemoglobinu a nejsou přítomny žádné klinické příznaky. Vyskytuje se v důsledku deleční nebo nedeleční mutace.[1] | - α / α α | |||||
| Dva |  Krvetvorba (tvorba krevních buněk) Tento stav se nazývá znak alfa talasémie; dva α geny umožňují téměř normální tvorba červených krvinek, ale mírný mikrocytární hypochromní anémie je viděn. Tuto chorobu lze zaměnit za anémie z nedostatku železa a nevhodně ošetřené železem.[3][1] Znak alfa-talasemie může existovat ve dvou formách:[1]
| - - / α α nebo - α / - α | |||||
| Tři | Tato podmínka se nazývá nemoc hemoglobinu H.; v krvi jsou přítomny dva nestabilní hemoglobiny; hemoglobin Barts (tetramerní γ řetězce ) a hemoglobin H (tetramerický β řetězce ). Oba tyto nestabilní hemoglobiny mají vyšší afinitu ke kyslíku než normální hemoglobin.[14] Mikrocytární hypochromní anémie s cílové buňky a Heinzova těla (vysrážená HbH) na nátěr periferní krve může nastat, stejně jako hepatosplenomegalie. Toto onemocnění je pozorováno v dětství nebo v raném dospělém životě; anémie a hepatosplenomegalie.[nutná lékařská citace ] | - - / - α | |||||
| Čtyři | Toto je známé jako alfa thalassemia major; tyto plody jsou edematózní, mají málo cirkulujícího hemoglobinu a přítomný hemoglobin jsou všechny tetramerní γ řetězce. Když jsou ovlivněny všechny čtyři alely, plod pravděpodobně nepřežije těhotenství bez in utero zásah; většina kojenců s alfa-thalassemií major se narodila mrtvá hydrops fetalis. Plody ošetřené nitroděložní transfuze po celou dobu těhotenství počínající v raném gestačním věku může přežít až do narození s přijatelnou nemocností. Po narození zahrnují možnosti léčby transplantaci kostní dřeně nebo pokračující chronické transfuze.[15] | - -/- - | |||||
| α α / α α = normální: „α α“ před „/“ představuje jeden chromozom a „α α“ za „/“ jeho homologní chromozom. | |||||||
Laboratorní diagnostika
Počáteční laboratorní diagnóza by měla zahrnovat a kompletní krevní obraz a indexy červených krvinek.[8] Také, a nátěr periferní krve by měl být pečlivě zkontrolován.[8]
Analýza hemoglobinu je důležitá pro diagnostiku alfa-talasémie, protože určuje typy a procenta přítomných typů hemoglobinu.[16] Existuje několik různých metod analýzy hemoglobinu, včetně hemoglobinová elektroforéza, kapilární elektroforéza a vysoce účinná kapalinová chromatografie.[16]
Molekulární analýza sekvencí DNA (Analýza DNA ) lze použít k potvrzení diagnózy alfa-talasémie, zejména k detekci nosičů alfa-talasémie (delece nebo mutace pouze u jedné nebo dvou alfa-globin geny).[16]
Léčba
Léčba alfa-talasémie může zahrnovat krevní transfuze udržovat hemoglobin na úrovni, která snižuje příznaky anémie. Rozhodnutí zahájit transfuzi závisí na klinické závažnosti onemocnění.[17] Splenektomie je možnou možností léčby ke zvýšení hladin celkového hemoglobinu v případě zhoršení anémie způsobené hyperaktivní nebo zvětšenou slezinou, nebo pokud není možná transfuzní léčba.[18] Splenektomii se však vyhýbáme, pokud jsou k dispozici jiné možnosti kvůli zvýšenému riziku závažných infekcí a trombóza.[18]
Dodatečně, žlučové kameny může být problém, který by vyžadoval chirurgický zákrok. Sekundární komplikace z febrilní epizoda by měla být sledována a většina jedinců žije bez nutnosti léčby.[1][4]
Dodatečně, transplantace kmenových buněk by měla být považována za léčbu (a léčbu), která se nejlépe provádí v raném věku. Další možnosti, jako např genová terapie, se stále vyvíjejí.[19]
Studie Kregera et al., Která kombinuje retrospektivní přehled tří případů alfa talasémie major a literární přehled 17 případů, zjistila, že transfuze dělohy může vést k příznivým výsledkům. Úspěšná transplantace hematopoetických buněk byla nakonec provedena u čtyř pacientů.[20]
Epidemiologie

Celosvětová distribuce zděděné alfa-talasémie odpovídá oblastem malárie expozice, což naznačuje ochrannou roli. Alfa-talasémie je tedy v subsaharské Africe běžná Afrika, Středomořská pánev a obecně tropické (a subtropické) oblasti. Epidemiologie alfa-talasémie v USA odráží tento globální distribuční vzorec. Přesněji řečeno, onemocnění HbH je vidět u Jihovýchodní Asie a střední východ, zatímco Hb Bart hydrops fetalis je uznáván pouze v jihovýchodní Asii.[21]Údaje naznačují, že 15% řecký a Turečtí Kypřané jsou dopravci beta-talasémie geny, zatímco 10% populace nese geny pro alfa-talasemii.[22]
Viz také
Reference
- ^ A b C d E F G h Origa, Raffaella; Moi, Paolo; Galanello, Renzo; Cao, Antonio (1. ledna 1993). "Alfa-talasémie". GeneReviews. PMID 20301608. Citováno 22. září 2016.aktualizace 2013
- ^ A b BRS Patologie (4. vydání). Lippincott Williams & Wilkins lékařský. Prosince 2009. str. 162. ISBN 978-1451115871.
- ^ A b C d E „Zpracování alfa talasemie: Aspekty přístupu, laboratorní studie, elektroforéza hemoglobinu“. emedicine.medscape.com. Citováno 24. května 2016.
- ^ A b "Komplikace a léčba | Talasemie | Poruchy krve | NCBDDD | CDC". www.cdc.gov. Citováno 22. září 2016.
- ^ Online Mendelian Inheritance in Man (OMIM): Hemoglobin - alfa lokus 1; HBA1 - 141800
- ^ Online Mendelian Inheritance in Man (OMIM): Hemoglobin - alfa lokus 2; HBA2 - 141850
- ^ Lanzkowského příručka pediatrické hematologie a onkologie 6. vydání (2016).
- ^ A b C d E "Alfa-talasémie - příznaky, diagnostika a léčba | Osvědčené postupy BMJ". bestpractice.bmj.com. Citováno 17. listopadu 2019.
- ^ Reference, Genetics Home. "Alfa thalassemia". Genetická domácí reference. Citováno 25. listopadu 2019.
- ^ Origa, Raffaella; Moi, Paolo (1993), Adam, Margaret P .; Ardinger, Holly H .; Pagon, Roberta A .; Wallace, Stephanie E. (eds.), "Alfa-talasémie", GeneReviews®University of Washington, Seattle, PMID 20301608, vyvoláno 25. listopadu 2019
- ^ "Hodnocení anémie - etiologie | Osvědčené postupy BMJ". bestpractice.bmj.com. Citováno 25. listopadu 2019.
- ^ Steensma DP, Gibbons RJ, Higgs DR (leden 2005). „Získaná alfa-talasémie ve spojení s myelodysplastickým syndromem a jinými hematologickými malignitami“. Krev. 105 (2): 443–52. doi:10.1182 / krev-2004-07-2792. PMID 15358626.
- ^ A b Galanello R, Cao A (únor 2011). "Kontrola genových testů. Alfa-talasémie". Genetika v medicíně. 13 (2): 83–8. doi:10.1097 / GIM.0b013e3181fcb468. PMID 21381239.
- ^ "Hemoglobin H nemoc". Orphanet. Citováno 22. září 2016.
- ^ Vichinsky EP (1. ledna 2009). „Alfa thalassemia major - nové mutace, nitroděložní léčba a výsledky“. Hematologie. Americká společnost pro hematologii. Vzdělávací program. 2009 (1): 35–41. doi:10.1182 / asheducation-2009.1.35. PMID 20008180.
- ^ A b C Viprakasit, Vip; Ekwattanakit, Supachai (1. dubna 2018). "Klinická klasifikace, screening a diagnostika talasémie". Hematologické / onkologické kliniky Severní Ameriky. Talasémie. 32 (2): 193–211. doi:10.1016 / j.hoc.2017.11.006. ISSN 0889-8588. PMID 29458726.
- ^ „UpToDate“. www.uptodate.com. Citováno 25. listopadu 2019.
- ^ A b Taher, Ali; Musallam, Khaled; Cappellini, Maria Domenica, eds. (2017). Pokyny pro léčbu talasemie, která není závislá na transfuzích (NTDT) (2. vyd.). Mezinárodní nadace Thalassemia. str. 24–32. Citováno 5. listopadu 2019.
- ^ "Thalassemia | Lékař | Pacient". Trpěliví. Citováno 22. září 2016.
- ^ Kreger EM, Singer ST, Witt RG, Sweeters N, Lianoglou B, Lal A a kol. (Prosinec 2016). „Příznivé výsledky po transfuzi dělohy u plodů s alfa thalassemia major: série případů a přehled literatury“. Prenatální diagnostika. 36 (13): 1242–1249. doi:10,1002 / pd.4966. PMID 27862048.
- ^ Harteveld CL, Higgs DR (květen 2010). "Alfa-talasémie". Orphanet Journal of Rare Diseases. 5 (1): 13. doi:10.1186/1750-1172-5-13. PMC 2887799. PMID 20507641.
- ^ Snadná hematologie. AuthorHouse. 06.02.2013. ISBN 9781477246511.strana 246
Další čtení
- Anie KA, Massaglia P (březen 2014). „Psychologické terapie pro talasemii“. Cochrane Database of Systematic Reviews (3): CD002890. doi:10.1002 / 14651858.cd002890.pub2. PMC 7138048. PMID 24604627.
- Galanello R, Cao A (únor 2011). "Kontrola genových testů. Alfa-talasémie". Genetika v medicíně. 13 (2): 83–8. doi:10.1097 / GIM.0b013e3181fcb468. PMID 21381239.
externí odkazy
- „Co jsou Thalassemias? - NHLBI, NIH“. www.nhlbi.nih.gov. Citováno 15. září 2016.
| Klasifikace | |
|---|---|
| Externí zdroje |
| Scholia má téma profil pro Alfa-talasémie. |