HBB - HBB


Beta globin (označovaný také jako HBB, β-globin, hemoglobin beta, hemoglobin betanebo nejlépe hemoglobinová podjednotka beta) je globin protein, který spolu s alfa globinem (HBA ), tvoří nejběžnější formu hemoglobin u dospělých lidí HbA.[4] Je dlouhý 147 aminokyselin a má molekulovou hmotnost 15 867 Da. Normální dospělý člověk HbA je heterotetramer skládající se ze dvou alfa řetězců a dvou beta řetězců.
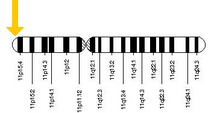
HBB je kódován HBB gen zapnutý lidský chromozom 11. Mutace v genu produkují několik variant proteinů, které jsou spojeny s genetickými poruchami, jako je srpkovitá nemoc a beta talasémie, stejně jako prospěšné vlastnosti, jako je genetická odolnost vůči malárii.[5][6]
Genový lokus
Protein HBB je produkován genem HBB který se nachází v multigenním místě Místo β-globinu na chromozom 11, konkrétně na pozici krátkého ramene 15.4. Výraz beta globinu a sousedních globinů v lokusu β-globinu je řízen jediným oblast kontroly lokusu (LCR), nejdůležitější regulační prvek v lokusu umístěném před geny globinu.[7] Normální alelická varianta je 1600 základní páry (bp) dlouhý a obsahuje tři exony. Pořadí genů v klastru beta-globinu je 5 '- epsilon – gamma-G – gama-A – delta - beta - 3 '.[4]
Interakce
HBB interaguje s Hemoglobin, alfa 1 (HBA1) za vzniku hemoglobinu A, hlavního hemoglobinu u dospělých lidí.[8][9] Interakce je dvojí. Nejprve se jeden HBB a jeden HBA1 nekovalentně spojí a vytvoří dimer. Za druhé, dva dimery se spojí a vytvoří čtyřřetězcový tetramer, který se stane funkčním hemolglobinem.[10]
Přidružené genetické poruchy
Beta talasémie
Beta talasémie je zděděná genetická mutace v jedné (Beta thalassemia minor) nebo v obou (Beta thalassemia major) alel beta globinu na chromozomu 11. Mutantní alely jsou dále rozděleny do dvou skupin: β0, ve které není vytvořen žádný funkční β-globin, a β +, ve kterém se produkuje malé množství normálního proteinu β-globinu. Beta thalassemia minor se vyskytuje, když jedinec zdědí jednu normální alelu Beta a jednu abnormální alelu Beta (buď β0, nebo β +). Beta thalassemia minor má za následek mírnou mikrocytární anémii, která je často asymptomatická nebo může způsobit únavu nebo bledou pokožku. Beta thalassemia major nastává, když člověk zdědí dvě abnormální alely. Může to být buď dvě alely β +, dvě alely β0, nebo každá z nich. Beta thalassemia major je vážný zdravotní stav. Těžká anémie se objevuje od 6 měsíců věku. Bez lékařského ošetření často dochází k smrti před dosažením věku 12 let. [11] Beta thalassemia major lze léčit celoživotně krevní transfuze nebo transplantace kostní dřeně.[12][13]
Podle nedávné studie mutace stop gain Gln40stop v HBB gen je běžnou příčinou autozomálně recesivní Beta-talasémie v Sardinští lidé (téměř exkluzivní na Sardinii). Nositelé této mutace vykazují zvýšený počet červených krvinek. Z kuriozity byla stejná mutace také spojena s poklesem séra LDL úrovně u dopravců, takže autoři naznačují, že je to kvůli potřebě cholesterol regenerovat buněčné membrány.[14]
Srpkovitá nemoc
Více než tisíc přirozeně se vyskytujících HBB byly objeveny varianty. Nejběžnější je HbS, který způsobuje srpkovitá nemoc. HbS je produkován a bodová mutace v HBB ve kterém kodon GAG je nahrazen GTG. To má za následek nahrazení hydrofilní aminokyseliny kyselina glutamová s hydrofobní aminokyselinou valin na šesté pozici (β6Glu → Val). Tato substituce vytváří hydrofobní místo na vnější straně proteinu, které se lepí na hydrofobní oblast beta řetězce sousední molekuly hemoglobinu. To dále způsobuje shlukování molekul HbS do tuhých vláken, což způsobuje „srpkování“ celku červené krvinky v homozygotní (HbS / HbS) podmínka.[15] Homozygotní alela se stala jedním z nejsmrtelnějších genetických faktorů,[16] zatímco lidé heterozygotní pro mutovanou alelu (HbS / HbA) jsou odolné vůči malárie a vyvinout minimální účinky anémie.[17]
Hemoglobin C.
Nemoc srpkovitých buněk úzce souvisí s dalším zvaným mutantním hemoglobinem hemoglobin C. (HbC), protože je lze zdědit společně.[18] Mutace HbC je ve stejné poloze v HbS, ale kyselina glutamová je nahrazena lysin (β6Glu → Lys). Mutace je zvláště rozšířená v západoafrických populacích. HbC poskytuje téměř plnou ochranu proti Plasmodium falciparum u homozygotních (CC) jedinců a střední ochrana u heterozygotních (AC) jedinců.[19] To naznačuje, že HbC má silnější vliv než HbS a předpokládá se, že nahradí HbS v endemických oblastech s malárií.[20]
Hemoglobin E
Další bodová mutace v HBB, ve které je kyselina glutamová nahrazena lysinem v poloze 26 (β26Glu → Lys), vede k tvorbě hemoglobin E (HbE).[21] HbE má velmi nestabilní asociaci α- a β-globin. I když má nestabilní protein sám o sobě mírný účinek, zděděný s vlastnostmi HbS a thalassemie, stává se z něj život ohrožující forma β-thalassemie. Mutace má relativně nedávný původ, což naznačuje, že byla výsledkem selektivního tlaku proti závažné malárii falciparum, protože heterozygotní alela brání rozvoji malárie.[22]
Lidská evoluce
Malárie kvůli Plasmodium falciparum je hlavním selektivním faktorem v lidská evoluce.[6][23] Ovlivnilo to mutace v HBB v různé míře, což vede k existenci mnoha variant HBB. Některé z těchto mutací nejsou přímo smrtelné a místo toho propůjčují rezistenci vůči malárii, zejména v Africe, kde je malárie epidemická.[24] U lidí afrického původu se vyvinula vyšší míra mutované HBB, protože heterozygotní jedinci mají deformovanou červenou krvinku, která brání útokům malarických parazitů. Mutanty HBB jsou tedy zdrojem pozitivní selekce v těchto oblastech a jsou důležité pro jejich dlouhodobé přežití.[5][25] Takové selekční markery jsou důležité pro sledování lidských předků a diverzifikace z Afriky.[26]
Viz také
Reference
- ^ A b C GRCh38: Vydání souboru 89: ENSG00000244734 - Ensembl, Květen 2017
- ^ „Human PubMed Reference:“. Národní centrum pro biotechnologické informace, Americká národní lékařská knihovna.
- ^ „Myš PubMed Reference:“. Národní centrum pro biotechnologické informace, Americká národní lékařská knihovna.
- ^ A b „Entrez Gene: HBB hemoglobin, beta“.
- ^ A b Sabeti, Pardis C (2008). „Přirozený výběr: odhalování mechanismů evoluční adaptace na infekční onemocnění“. Přírodní výchova. 1 (1): 13.
- ^ A b Kwiatkowski DP (2005). „Jak malárie ovlivnila lidský genom a co nás lidská genetika může o malárii naučit“. American Journal of Human Genetics. 77 (2): 171–192. doi:10.1086/432519. PMC 1224522. PMID 16001361.
- ^ Levings PP, Bungert J (2002). "Oblast kontroly lidského beta-globinového lokusu". Eur. J. Biochem. 269 (6): 1589–99. doi:10.1046 / j.1432-1327.2002.02797.x. PMID 11895428.
- ^ Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, Timm J, Mintzlaff S, Abraham C, Bock N, Kietzmann S, Goedde A, Toksöz E , Droege A, Krobitsch S, Korn B, Birchmeier W, Lehrach H, Wanker EE (2005). „Síť interakce lidský protein-protein: zdroj pro anotování proteomu“. Buňka. 122 (6): 957–968. doi:10.1016 / j.cell.2005.08.029. hdl:11858 / 00-001M-0000-0010-8592-0. PMID 16169070. S2CID 8235923.
- ^ Shaanan B (1983). "Struktura lidského oxyhemoglobinu při rozlišení 2,1 A". J. Mol. Biol. ANGLIE. 171 (1): 31–59. doi:10.1016 / S0022-2836 (83) 80313-1. ISSN 0022-2836. PMID 6644819.
- ^ "Syntéza hemoglobinu". harvard.edu. Harvardská Univerzita. 2002. Citováno 18. listopadu 2014.
- ^ H. Franklin Bunn; Vijay G. Sankaran (2017). „8“. Patologie krevních poruch. 927–933.
- ^ Muncie HL, Campbell J (2009). "Alfa a beta talasémie". Americký rodinný lékař. 80 (4): 339–44. PMID 19678601.
- ^ „Beta thalassemia“. Genetická domácí reference. Americká národní lékařská knihovna. 11. listopadu 2014. Citováno 18. listopadu 2014.
- ^ Sidore, C .; et al. (2015). „Sekvenování genomu objasňuje sardinskou genetickou architekturu a rozšiřuje asociační analýzy lipidových a krevních zánětlivých markerů“. Genetika přírody. 47 (11): 1272–1281. doi:10,1038 / ng.3368. PMC 4627508. PMID 26366554.
- ^ Thom CS, Dickson CF, Gell DA, Weiss MJ (2013). „Varianty hemoglobinu: biochemické vlastnosti a klinické koreláty“. Cold Spring Harb Perspect Med. 3 (3): a011858. doi:10.1101 / cshperspect.a011858. PMC 3579210. PMID 23388674.
- ^ Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker- Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M , Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V , Flaxman AD, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez-Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ , Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaram an S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo JP, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL, Mabweijano J, MacIntyre MF, Mallinger L, březen L , Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGrath J, Mensah GA, Merriman TR, Michaud C, Miller M, Miller TR, Mock C, Mocumbi AO, Mokdad AA, Moran A, Mulholland K, Nair MN, Naldi L, Narayan KM, Nasseri K, Norman P, O'Donnell M, Omer SB, Ortblad K, Osborne R, Ozgediz D, Pahari B, Pandian JD, Rivero AP, Padilla RP, Perez -Ruiz F, Perico N, Phillips D, Pierce K, Pope CA, Porrini E, Pourmalek F, Raju M, Ranganathan D, Rehm JT, Rein DB, Remuzzi G, Rivara FP, Roberts T, De León FR, Rosenfeld LC, Rushton L, Sacco RL, Salomon JA, Sampson U, Sanman E, Schwebel DC, Segui-Gomez M, Shepard DS, Singh D, Singleton J, Sliwa K, Smith E, řízení A, Taylor JA, Thomas B, Tleyjeh IM, Towbin JA, Truelsen T, Undurraga EA, Venketasubramanian N, Vijayakumar L, Vos T, Wagne r GR, Wang M, Wang W, Watt K, Weinstock MA, Weintraub R, Wilkinson JD, Woolf AD, Wulf S, Yeh PH, Yip P, Zabetian A, Zheng ZJ, Lopez AD, Murray CJ, AlMazroa MA, Memish ZA (2012). „Globální a regionální úmrtnost z 235 příčin úmrtí u 20 věkových skupin v letech 1990 a 2010: systematická analýza studie Global Burden of Disease Study 2010“. Lanceta. 380 (9859): 2095–128. doi:10.1016 / S0140-6736 (12) 61728-0. hdl:10536 / DRO / DU: 30050819. PMID 23245604. S2CID 1541253.
- ^ Luzzatto L (2012). "Srpkovitá anémie a malárie". Mediterr J Hematol Infect Dis. 4 (1): e2012065. doi:10.4084 / MJHID.2012.065. PMC 3499995. PMID 23170194.
- ^ Piel FB, Howes RE, Patil AP, Nyangiri OA, Gething PW, Bhatt S, Williams TN, Weatherall DJ, Hay SI (2013). „Distribuce hemoglobinu C a jeho prevalence u novorozenců v Africe“. Vědecké zprávy. 3 (1671): 1671. Bibcode:2013NatSR ... 3E1671P. doi:10.1038 / srep01671. PMC 3628164. PMID 23591685.
- ^ Modiano D, Luoni G, Sirima BS, Simporé J, Verra F, Konaté A, Rastrelli E, Olivieri A, Calissano C, Paganotti GM, D'Urbano L, Sanou I, Sawadogo A, Modiano G, Coluzzi M (2001). „Hemoglobin C chrání před klinickou malárií Plasmodium falciparum“. Příroda. 414 (6861): 305–308. Bibcode:2001 Natur.414..305M. doi:10.1038/35104556. PMID 11713529. S2CID 4360808.
- ^ Verra F, Bancone G, Avellino P, Blot I, Simporé J, Modiano D (2007). „Hemoglobin C a S v přirozeném výběru proti malárii Plasmodium falciparum: nepřeberné množství nebo jediný společný adaptivní mechanismus?“. Parassitologia. 49 (4): 209–13. PMID 18689228.
- ^ Olivieri NF, Pakbaz Z, Vichinsky E (2011). „Hb E / beta-talasémie: běžná a klinicky různorodá porucha“. Indický žurnál lékařského výzkumu. 134 (4): 522–531. PMC 3237252. PMID 22089616.
- ^ Chotivanich K, Udomsangpetch R, Pattanapanyasat K, Chierakul W, Simpson J, Looareesuwan S, White N (2002). „Hemoglobin E: vyvážený polymorfismus chránící před vysokými parazitemiemi, a tedy závažný P falciparum malárie". Krev. 100 (4): 1172–1176. doi:10.1182 / krev.V100.4.1172.h81602001172_1172_1176. PMID 12149194.
- ^ Verra F, Mangano VD, Modiano D (2009). „Genetika citlivosti na Plasmodium falciparum: od klasických genů rezistence na malárii po genomové asociační studie“. Parazitická imunologie. 31 (5): 234–53. doi:10.1111 / j.1365-3024.2009.01106.x. PMID 19388945. S2CID 23734166.
- ^ Tishkoff SA, Williams SM (2002). "Genetická analýza afrických populací: vývoj člověka a komplexní nemoc". Genetika hodnocení přírody. 3 (8): 611–21. doi:10.1038 / nrg865. PMID 12154384. S2CID 7801737.
- ^ Excoffier L (2002). „Demografická historie člověka: zdokonalení nedávného modelu afrického původu“. Aktuální názor na genetiku a vývoj. 12 (6): 675–682. doi:10.1016 / S0959-437X (02) 00350-7. PMID 12433581.
- ^ Reed FA, Tishkoff SA (2006). „Africká lidská rozmanitost, původ a migrace“. Aktuální názor na genetiku a vývoj. 16 (6): 597–605. doi:10.1016 / j.gde.2006.10.008. PMID 17056248.
Další čtení
- Higgs DR, Vickers MA, Wilkie AO, Pretorius IM, Jarman AP, Weatherall DJ (1989). „Přehled molekulární genetiky lidského klastru genů alfa-globinů“. Krev. 73 (5): 1081–104. doi:10.1182 / krev. V73.5.1081.1081. PMID 2649166.
- Giardina B, Messana I, Scatena R, Castagnola M (1995). "Více funkcí hemoglobinu". Krit. Biochem. Mol. Biol. 30 (3): 165–96. doi:10.3109/10409239509085142. PMID 7555018.
- Salzano AM, Carbone V, Pagano L, Buffardi S, De RC, Pucci P (2002). „Hb Vila Real [beta36 (C2) Pro -> His] v Itálii: charakterizace aminokyselinové substituce a mutace DNA“. Hemoglobin. 26 (1): 21–31. doi:10.1081 / HEM-120002937. PMID 11939509. S2CID 40757080.
- Frischknecht H, Dutly F (2007). „Duplikace / inzerce 65 bp v exonu II genu beta globinu způsobující beta0-talasemii“. Haematologica. 92 (3): 423–4. doi:10,3324 / haematol.10785. PMID 17339197.
externí odkazy
- Přehled všech strukturálních informací dostupných v PDB pro UniProt: P68871 (Lidská hemoglobinová podjednotka beta) na PDBe-KB.
- Přehled všech strukturálních informací dostupných v PDB pro UniProt: P02088 (Myší hemoglobinová podjednotka beta-1) na PDBe-KB.
Galerie PDB | |
|---|---|
|