Talasémie - Thalassemia
| Talasémie | |
|---|---|
| Ostatní jména | Thalassemia, středomořská anémie |
 | |
| Film z periferní krve od osoby s Delta Beta thalassemií | |
| Výslovnost | |
| Specialita | Hematologie |
| Příznaky | Pocit unavený, bledá kůže, zvětšená slezina, nažloutlá kůže, tmavá moč[1] |
| Příčiny | Genetický (autozomálně recesivní )[2] |
| Diagnostická metoda | Krevní testy, genetické testy[3] |
| Léčba | Krevní transfuze, chelatace železa, kyselina listová[4] |
| Frekvence | 280 milionů (2015)[5] |
| Úmrtí | 16,800 (2015)[6] |
Thalassemias jsou zděděni poruchy krve charakterizované snížením hemoglobin Výroba.[7] Příznaky závisí na typu a mohou se lišit od žádných po závažné.[1] Často je mírné až těžké anémie (nízký červené krvinky nebo hemoglobin).[1] Anémie může vést k pocitu unavený a bledá kůže.[1] Mohou také nastat problémy s kostmi, zvětšená slezina, nažloutlá kůže a tmavá moč.[1] U dětí může dojít k pomalému růstu.[1]
Thalassemias jsou genetické poruchy zdědil od rodičů člověka.[2] Existují dva hlavní typy, alfa talasémie a beta talasémie.[7] Závažnost alfa a beta talasémie závisí na tom, pro kolik ze čtyř genů alfa globin nebo dva geny pro beta globin chybí.[2] Diagnóza je obvykle pomocí krevních testů včetně a kompletní krevní obraz, speciální testy na hemoglobin a genetické testy.[3] Diagnóza může nastat před narozením prenatální testování.[8]
Léčba závisí na typu a závažnosti.[4] Léčba pacientů s těžším onemocněním často zahrnuje pravidelnou léčbu krevní transfuze, chelatace železa, a kyselina listová.[4] Chelataci železem lze provést deferoxamin, deferasirox nebo deferipron.[4][9] Občas, a transplantace kostní dřeně může být možnost.[4] Komplikace mohou zahrnovat přetížení železem z transfuzí s výsledkem srdce nebo nemoc jater, infekce, a osteoporóza.[1] Pokud slezina se příliš zvětší, chirurgické odstranění může být vyžadováno.[1] Pacienti s talasemií, kteří nereagují dobře na krevní transfuze, mohou užívat hydroxymočovinu nebo thalidomid a někdy i kombinaci obou.[10] Hydroxymočovina je jediným lékem schváleným FDA pro talasémii. Pacienti, kteří užívali 10 mg / kg hydroxymočoviny každý rok po dobu jednoho roku, měli významně vyšší hladiny hemoglobinu a byla to dobře tolerovaná léčba u pacientů, kteří nereagovali dobře na krevní transfuze.[11] Další induktor hemoglobinu zahrnuje thalidomid, i když nebyl testován v klinickém prostředí. Kombinace thalidomidu a hydroxymočoviny vedla k významnému zvýšení hladin hemoglobinu u pacientů závislých na transfúzi a na transfuzích nezávislých [12]
Od roku 2015 se talasemie vyskytuje u přibližně 280 milionů lidí, přičemž přibližně 439 000 má závažné onemocnění.[13] Je to nejčastější u lidí z Itálie, Řecka, Středního východu, Jihoasijský a afrického původu.[7] Muži a ženy mají podobnou míru onemocnění.[14] V roce 2015 to vedlo k 16 800 úmrtím, což je pokles z 36 000 úmrtí v roce 1990.[6][15] Ti, kteří mají menší stupně talasemie, podobné těm s srpkovitá vlastnost, mít určitou ochranu proti malárie vysvětlující, proč jsou častější v oblastech světa, kde existuje malárie.[16]
Příznaky a symptomy

- Přetížení železa: Lidé s talasemií mohou mít v těle přetížení železem, a to buď z důvodu samotné nemoci, nebo z častých transfuzí krve. Příliš mnoho železa může mít za následek poškození srdce, jater a endokrinní systém, což zahrnuje žlázy, které produkují hormony, které regulují procesy v celém těle. Poškození je charakterizováno nadměrnými usazeninami železa. Bez adekvátní terapie chelatací železa akumulují téměř všichni pacienti s beta-thalasemií potenciálně smrtelné hladiny železa.[17]
- Infekce: Lidé s talasemií mají zvýšené riziko infekce. To platí zejména v případě, že byla odstraněna slezina.[18]
- Kostní deformity: Talasemie může způsobit rozšíření kostní dřeně, což způsobí rozšíření kostí. To může mít za následek abnormální strukturu kostí, zejména obličeje a lebky. Expanze kostní dřeně také činí kosti tenkými a křehkými, což zvyšuje riziko zlomenin.[19]
- Zvětšená slezina: Slezina pomáhá v boji proti infekci a filtruje nežádoucí materiál, jako jsou staré nebo poškozené krvinky. Thalassemie je často doprovázena destrukcí velkého počtu červených krvinek a odstranění těchto buněk způsobuje zvětšení sleziny. Splenomegalie může anémii zhoršit a snížit životnost transfuzovaných červených krvinek. Silné zvětšení sleziny může vyžadovat její odstranění.[20]
- Zpomalené tempo růstu: anémie může způsobit zpomalení růstu dítěte. Puberta může být také zpožděna u dětí s talasemií.[21]
- Srdeční problémy: Nemoci, jako je městnavé srdeční selhání a abnormální srdeční rytmus, mohou být spojeny s těžkou talasemií.[22]
Způsobit
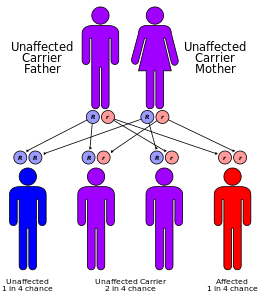
Jak α-, tak β-thalassemie se často dědí v autosomální recesivní způsob. Případy dominantně byly hlášeny zděděné α- a β-thalassemie, z nichž první byla v irské rodině se dvěma delecemi 4 a 11 bp v exonu 3 přerušenými vložením 5 bp do genu β-globinu. U autozomálně recesivních forem onemocnění musí být oba rodiče nositeli dítěte, které má být postiženo. Pokud oba rodiče nesou znak hemoglobinopatie, riziko pro postižené dítě je 25% za každé těhotenství.
Vývoj
Mít jedinou genetickou variantu pro talasemii může chránit před malárií a proto může být výhodou.[23]
Lidé s diagnózou heterozygotní (nosič) β-thalasémie mají určitou ochranu proti ischemická choroba srdeční.[24]
Patofyziologie
Normálně je většina dospělého hemoglobinu (HbA ) se skládá ze čtyř proteinových řetězců, dvou α a dvou β-globinových řetězců uspořádaných do a heterotetramer. U talasemie mají pacienti poruchy buď v α nebo β-globinovém řetězci, které způsobují tvorbu abnormálních červených krvinek (v srpkovitá nemoc, což je hemoglobinopatie a nikoli správná talasémie, je mutace specifická pro β-globin).
Talasemie se klasifikují podle toho, který řetězec molekuly hemoglobinu je ovlivněn. v α-thalassemias, je ovlivněna produkce řetězce α-globinu, zatímco v β-talasémie, je ovlivněna produkce β-globinového řetězce.
P-globinové řetězce jsou kódovány jediným genem chromozom 11; α-globinové řetězce jsou kódovány dvěma úzce spojenými geny chromozom 16.[25] U normální osoby se dvěma kopiemi každého chromozomu tedy dva lokusy kódují β řetězec a čtyři lokusy kódují α řetězec. Odstranění jednoho z α lokusů má vysokou prevalenci u lidí afrického nebo asijského původu, což zvyšuje jejich pravděpodobnost vývoje α-talasémie. β-thalassemie jsou nejen běžné v Afričané, ale také v Řekové a Italové.
Alfa-thalassemias
Α-thalassemias zahrnují geny HBA1[26] a HBA2,[27] zděděný v a Mendelian recesivní móda. Dva genové loci a tak existují čtyři alely. Existují dva genetické lokusy pro a-globin, tedy čtyři alely jsou v diploidních buňkách. Dvě alely jsou mateřské a dvě alely jsou otcovského původu. Závažnost α-thalassemií koreluje s počtem postižených α-globinů; alely: čím větší, tím závažnější budou projevy nemoci.[28] Alfa-thalassemie vedou ke snížení produkce alfa-globinu; proto se produkuje méně alfa-globinových řetězců, což má za následek přebytek β řetězců u dospělých a přebytek γ řetězců u novorozenců. Přebytek β řetězců tvoří nestabilní tetramery (nazývané hemoglobin H nebo HbH 4 beta řetězců), které mají abnormální křivky disociace kyslíku. Alfa thalassemias se často vyskytují u lidí z jihovýchodní Asie, Středního východu, Číny a u lidí afrického původu.[29]
| # chybějících alel | Druhy alfa talasémie[28] | Příznaky |
|---|---|---|
| 1 | Tichý nosič | Žádné příznaky |
| 2 | Znak alfa talasemie | Drobná anémie |
| 3 | Hemoglobin H nemoc | Mírná až střední anémie může vést normální život |
| 4 | Hydrops fetalis | K úmrtí plodu obvykle dochází při narození |
Beta-talasémie
Beta thalassemie jsou způsobeny mutacemi v Gen HBB na chromozomu 11,[30] také zděděné autozomálně, recesivně. Závažnost onemocnění závisí na povaze mutace a na přítomnosti mutací v jedné nebo obou alelách.
Mutované alely se nazývají β+ když je zachována částečná funkce (buď má protein sníženou funkci, nebo funguje normálně, ale je produkován ve sníženém množství) nebo βÓ, když není produkován žádný funkční protein.
Klinický obraz určuje situace obou alel:
- β thalassemia major (Středomoří anémie nebo Cooley anémie) je způsobena βÓ/ βÓ genotyp. Neprodukují se žádné funkční β řetězce, a proto nelze sestavit žádný hemoglobin A. Toto je nejtěžší forma β-talasémie;
- β thalassemia intermedia je způsobena β+/ βÓ nebo β+/ β+ genotyp. V této formě se produkuje určitý hemoglobin A;
- β thalassemia minor je způsobena β / βÓ nebo β / β+ genotyp. Pouze jedna ze dvou alel β globinu obsahuje mutaci, takže produkce β řetězce není nijak zvlášť ohrožena a pacienti mohou být relativně bez příznaků.
Beta talasemie se nejčastěji vyskytuje u lidí středomořského původu. V menší míře mohou být ovlivněni Číňané, ostatní Asiaté a Afroameričané.[29]
Delta-talasémie
Kromě řetězců alfa a beta přítomných v hemoglobinu jsou asi 3% dospělého hemoglobinu vyrobeny z řetězců alfa a delta. Stejně jako u beta thalassemie mohou nastat mutace, které ovlivňují schopnost tohoto genu produkovat delta řetězce.[Citace je zapotřebí ]
Kombinované hemoglobinopatie
Thalassemia může koexistovat s jinými hemoglobinopatie. Nejběžnější z nich jsou:
- Hemoglobin E / talasémie: běžné v Kambodža, Thajsko a části Indie, je klinicky podobný β thalassemia major nebo thalassemia intermedia.[Citace je zapotřebí ]
- Hemoglobin S / talasémie: běžné v Afričan a Středomoří populace, je klinicky podobný srpkovitá anémie, s další funkcí splenomegalie.[Citace je zapotřebí ]
- Hemoglobin C. / talasémie: běžné v Středomoří a Afričan populace, hemoglobin C / βÓ thalassemie způsobuje středně těžkou hemolytickou anémii se splenomegalií; hemoglobin C / β+ thalassemia produkuje mírnější onemocnění.[Citace je zapotřebí ]
- Hemoglobin D / talasémie: běžné v severozápadních částech Indie a Pákistán (Region Paňdžáb ).[31]
Diagnóza
Thalassemii lze diagnostikovat pomocí a kompletní krevní obraz, hemoglobinová elektroforéza a testování DNA.[32]Hemoglobinová elektroforéza není v rozvojových zemích široce dostupná, proto lze pro diagnostiku talasémie použít také menterský index. Ačkoli to není diagnostický test, ale může poskytnout spravedlivou představu o možnosti talasémie. Mentzerův index lze vypočítat z úplné zprávy o krevním obrazu. K tomuto účelu lze také použít Mentzerovy indexové kalkulačky[33].
Prevence
The Americká vysoká škola porodníků a gynekologů doporučuje všem lidem, kteří uvažují o otěhotnění, nechat se otestovat, zda mají talasemii.[34] Genetické poradenství a genetické testování se doporučují rodinám, které nesou vlastnost talasemie.
Ve společnosti existuje politika prověřování Kypr snížit míru talasémie, která od zavedení programu v 70. letech (která zahrnuje také prenatální screening a potrat) snížila počet dětí narozených s tímto onemocněním z jednoho ze 158 porodů na téměř nulu.[35] Řecko má také screeningový program k identifikaci osob, které jsou přepravci.[36]
v Írán jako předmanželský screening se nejprve zkontrolují indexy červených krvinek u muže. Pokud ano mikrocytóza (střední buněčný hemoglobin <27 pg nebo střední objem červených krvinek <80 fl), žena je testována. Když jsou oba mikrocytární, jejich hemoglobin A2 koncentrace se měří. Pokud mají oba koncentraci vyšší než 3,5% (diagnostika znaku talasemie), jsou doporučeni místnímu místu určenému pro zdraví genetické poradenství.[37]
Ve městě se organizují rozsáhlé osvětové kampaně Indie[38] jak ze strany vládních, tak nevládních organizací ve prospěch dobrovolného předmanželského screeningu k detekci nositelů talasemie a manželství mezi oběma nositeli se důrazně nedoporučuje.
Řízení
Mírná talasemie: lidé s talasemií rysy po stanovení počáteční diagnózy nevyžadují lékařskou ani následnou péči.[39] Lidé s rysem β-talasémie by měli být varováni, že jejich stav může být chybně diagnostikován jako častější anémie z nedostatku železa. Měli by se vyhnout rutinnímu používání doplňky železa; nedostatek železa se může vyvinout během těhotenství nebo z chronického krvácení.[40] Poradenství je indikováno u všech osob s genetickými poruchami, zejména pokud je v rodině riziko těžké formy onemocnění, kterému lze předcházet.[41]
Anémie
Lidé s těžkou talasemií vyžadují lékařské ošetření. Režim krevní transfuze byl prvním měřítkem účinným při prodloužení života.[39]
Terapie růstovým hormonem
Existují určité důkazy substituční terapie růstovým hormonem může pomoci zvýšit rychlost růstu dětí s talasemií.[42]
Přetížení železa
Vícenásobné transfuze krve mohou vést k přetížení železem. Přetížení železem související s talasemií lze léčit pomocí chelatační terapie s léky deferoxamin, deferipron nebo deferasirox.[43] Tato léčba vedla ke zvýšení průměrné délky života u pacientů s thalassemia major.[43]
Deferoxamin je účinný pouze prostřednictvím denních injekcí, což ztěžuje jeho dlouhodobé užívání. Výhodou je nenákladnost a slušná dlouhodobá bezpečnost. Nežádoucí účinky jsou primární kožní reakce v okolí místa vpichu a ztráta sluchu.[43]
Výhodou přípravku Deferasirox je to, že je perorálním lékem. Mezi časté nežádoucí účinky patří: nevolnost, zvracení a průjem. Není však účinný u každého a pravděpodobně není vhodný u pacientů se závažnými srdečními problémy souvisejícími s přetížením železem. Náklady jsou také značné.[43]
Deferipron je lék, který se podává ústy. Nevolnost, zvracení a průjem jsou při jeho užívání poměrně časté.[43] Je k dispozici v Evropě i ve Spojených státech.[43][44] Zdá se, že je to nejúčinnější prostředek, když je srdce významně zapojeno.[43]
Neexistují žádné důkazy z randomizovaná kontrolovaná studie na podporu suplementace zinku při talasemii.[45]
Transplantace kostní dřeně
Transplantace kostní dřeně může nabídnout možnost léčby u mladých lidí, kteří mají HLA - neodpovídající dárce.[46] Míra úspěšnosti se pohybuje v rozmezí 80–90%.[46] Úmrtnost na postup je asi 3%.[47] Neexistují žádné randomizované kontrolované studie, které by testovaly bezpečnost a účinnost přípravku neidentický dárce transplantace kostní dřeně u osob s β-talasémií, kteří jsou závislí na transfuzi krve.[48]
Nemoci štěpu proti hostiteli (GvHD) jsou jedním relevantním vedlejším účinkem transplantace kostní dřeně. Je zapotřebí dalšího výzkumu k vyhodnocení, zda lze mezenchymální stromální buňky použít jako profylaxi nebo léčbu GvHD.[49]
Pokud daná osoba nemá kompatibilního dárce s HLA, lze použít jinou metodu zvanou transplantace kostní dřeně (BMT) z haploidentické matky na dítě (neodpovídající dárce). Ve studii s 31 lidmi byla míra přežití bez talasemie 70%, odmítnutí 23% a úmrtnost 7%. Nejlepší výsledky mají velmi mladí lidé.[50]
Epidemiologie
Beta forma talasémie je zvláště rozšířená mezi Středomoří národy a toto zeměpisné sdružení je odpovědné za svůj původní název.[51] Talasémie měla za následek 25 000 úmrtí v roce 2013, oproti 36 000 úmrtím v roce 1990.[15]
V Evropě se nejvyšší koncentrace choroby nacházejí v Řecko, pobřežní oblasti v krocan (zejména Egejská oblast jako Izmir, Balikesir, Aydin, Mugla, a Středomořský region jako Antalya, Adana, Mersin ), v částech Itálie, zejména jižní Itálie a dolní údolí Pádu. Hlavní středomořské ostrovy (kromě Baleáry ) jako Sicílie, Sardinie, Malta, Korsika, Kypr, a Kréta jsou obzvláště silně ovlivněny. Ostatní obyvatelé Středomoří, stejně jako lidé v blízkosti Středozemního moře, mají také vysokou míru talasemie, včetně lidí z Západní Asie a Severní Afrika. Daleko od Středomoří, Jižní Asiaté jsou také ovlivněny, přičemž nejvyšší koncentrace dopravců na světě (16–18% populace) je v Maledivy.[52]
V dnešní době se vyskytuje v populacích žijících v Africe, Severní a Jižní Americe a v USA Tharu lidé v Terai region Nepál a Indie.[53] Předpokládá se, že představuje mnohem nižší nemoci a úmrtí na malárii,[54] což odpovídá historické schopnosti Tharuse přežít v oblastech se silným napadením malárií, kde ostatní nemohli. Thalassemias jsou zvláště spojovány s lidmi středomořského původu, s Araby (zejména Palestinci a lidé palestinského původu) a Asiaté.[55] Na Maledivách je nejvyšší výskyt talasémie na světě s mírou přenosu 18% populace. Odhadovaná prevalence je 16% u lidí z Kypr, 1%[56] v Thajsko a 3–8% v populacích od Bangladéš, Čína, Indie, Malajsie a Pákistán. Thalassemias se vyskytují také u potomků lidí ze středomořských zemí (např. Řecko, Itálie, Španělsko a další), v Latinská Amerika.
Odhady naznačují, že přibližně 1,5% světové populace (80 - 90 milionů lidí) je nositelem β-talasémie.[57] Přesné údaje o sazbách dopravců v mnoha populacích však chybí, zejména v rozvojových oblastech světa, o nichž je známo nebo se očekává, že budou silně ovlivněny.[58][59] Vzhledem k prevalenci onemocnění v zemích s malými znalostmi talasemie může být obtížný přístup ke správné léčbě a diagnostice.[60] I když v rozvojových zemích existují některá diagnostická a léčebná zařízení, ve většině případů nejsou poskytována vládními službami a jsou k dispozici pouze pacientům, kteří si je mohou dovolit. Chudší populace mají obecně přístup pouze k omezeným diagnostickým zařízením spolu s transfuzí krve. V některých rozvojových zemích neexistují prakticky žádná zařízení pro diagnostiku nebo léčbu talasémie.[60]
Etymologie a synonymum
Slovo talasémie (/θ…lɪˈsiːmiə/) pochází z řecký thalassa (θάλασσα), "moře",[61] a Nová latina -émie (z řečtiny sloučenina stonek -aimia (-αιμία), z haima (αἷμα), „krev“).[62] Byl vytvořen, protože stav zvaný „středomořská anémie“ byl první popsáno u lidí z Středomoří etnik. „Středomořská anémie“ byla přejmenována thalassemia major jakmile byla genetika lépe pochopena. Slovo talasémie byl poprvé použit v roce 1932.[51]:877[63]
Společnost a kultura
V roce 2008 bylo ve Španělsku dítě selektivně implantován být lék na bratrovu talasemii. Dítě se narodilo z embrya, které bylo před implantací vyšetřeno na prosté choroby in vitro oplodnění. Zásoba dítěte imunologicky kompatibilní pupečníkové krve byla uložena na transplantaci jeho bratrovi. Transplantace byla považována za úspěšnou.[64] V roce 2009 skupina lékařů a specialistů v Chennai a Coimbatore zaregistroval úspěšnou léčbu talasemie u dítěte pomocí pupečníkové krve neovlivněného sourozence.[65]
Výzkum
Genová terapie
Genová terapie se zkoumá na talasemii.[66] Postup zahrnuje sběr hematopoetické kmenové buňky (HSC) z krve postižené osoby. HSC pak mají přidán gen beta-globinu pomocí a lentivirový vektor. Po zničení kostní dřeně postižené osoby dávkou chemoterapie (myeloablativní kondiční režim) se změněné HSC infundují zpět do postižené osoby, kde se v kostní dřeni zasadí, kde se množí. To potenciálně vede k postupnému zvyšování syntézy hemoglobinu A2 ve všech následujících vyvíjejících se červených krvinkách s výsledným ústupem anémie.[67]
Zatímco jedna osoba s beta talasémií již po léčbě v rámci výzkumné studie nevyžaduje krevní transfuze, nejedná se o schválenou léčbu od roku 2018.[66][68]
HbF indukce
Indikace HbF je pokusem o reaktivaci transkripce genu fetálního globinu.[69] Úsilí zahrnuje snahu narušit promotor genu pro fetální globin.[69]
Reference
- ^ A b C d E F G h „Jaké jsou příznaky a příznaky thalassemií?“. NHLBI. 3. července 2012. Archivováno z původního dne 16. září 2016. Citováno 5. září 2016.
- ^ A b C „Co způsobuje Thalassemias?“. NHLBI. 3. července 2012. Archivováno z původního dne 26. srpna 2016. Citováno 5. září 2016.
- ^ A b „Jak jsou diagnostikovány thalassemie?“. NHLBI. 3. července 2012. Archivováno z původního dne 16. září 2016. Citováno 5. září 2016.
- ^ A b C d E „Jak se zachází s Thalassemias?“. NHLBI. 3. července 2012. Archivováno z původního dne 16. září 2016. Citováno 5. září 2016.
- ^ GBD 2015 Incidence a prevalence nemocí a úrazů, spolupracovníci. (8. října 2016). „Globální, regionální a národní výskyt, prevalence a roky prožité se zdravotním postižením pro 310 nemocí a úrazů, 1990–2015: systematická analýza studie Global Burden of Disease Study 2015“. Lanceta. 388 (10053): 1545–1602. doi:10.1016 / S0140-6736 (16) 31678-6. PMC 5055577. PMID 27733282.
- ^ A b GBD 2015 Úmrtnost a příčiny smrti, spolupracovníci. (8. října 2016). „Globální, regionální a národní naděje dožití, úmrtnost ze všech příčin a specifická úmrtnost pro 249 příčin úmrtí, 1980–2015: systematická analýza studie Global Burden of Disease Study 2015“. Lanceta. 388 (10053): 1459–1544. doi:10.1016 / s0140-6736 (16) 31012-1. PMC 5388903. PMID 27733281.
- ^ A b C „Co jsou Thalassemias?“. NHLBI. 3. července 2012. Archivováno z původního dne 26. srpna 2016. Citováno 5. září 2016.
- ^ „Jak lze Thalassemiasovi zabránit?“. NHLBI. 3. července 2012. Archivováno z původního dne 16. září 2016. Citováno 5. září 2016.
- ^ "Chelatace železa". Citováno 15. července 2020.
- ^ Shah, Sandip; Sheth, Radhika; Shah, Kamlesh; Patel, Kinnari (únor 2020). „Bezpečnost a účinnost kombinace thalidomidu a hydroxymočoviny u β-thalassemia intermedia a major: retrospektivní pilotní studie“. British Journal of Hematology. 188 (3). doi:10.1111 / bjh.16272. ISSN 0007-1048.
- ^ Keikhaei, Bijan (2015). "Klinické a hematologické účinky hydroxymočoviny u pacientů s β-thalasemií intermediální". VĚSTNÍK KLINICKÉHO A DIAGNOSTICKÉHO VÝZKUMU. doi:10,7860 / JCDR / 2015 / 14807,6660.
- ^ Masera, Nicoletta; Tavecchia, Luisa; Capra, Marietta; Cazzaniga, Giovanni; Vimercati, Chiara; Pozzi, Lorena; Biondi, Andrea; Masera, Giuseppe (2010). „Optimální odpověď na thalidomid u pacienta s hlavní thalasemií rezistentní na konvenční terapii“. Krevní transfúze. doi:10.2450/2009.0102-09. ISSN 1723-2007. PMC 2809513. PMID 20104280.
- ^ Global Burden of Disease Study 2013, Collaborators (22. srpna 2015). „Globální, regionální a národní výskyt, prevalence a roky života s postižením pro 301 akutních a chronických onemocnění a úrazů ve 188 zemích, 1990–2013: systematická analýza studie Global Burden of Disease Study 2013“. Lanceta. 386 (9995): 743–800. doi:10.1016 / s0140-6736 (15) 60692-4. PMC 4561509. PMID 26063472.
- ^ Clin. Metody v Ped. Vydavatelé Jaypee Brothers. 2005. s. 21. ISBN 9788171798087.
- ^ A b GBD 2013 Úmrtnost a příčiny smrti, spolupracovníci (17. prosince 2014). „Globální, regionální a národní věková pohlaví specifická úmrtnost ze všech příčin a příčin specifická pro 240 příčin úmrtí, 1990–2013: systematická analýza studie Global Burden of Disease Study 2013“. Lanceta. 385 (9963): 117–71. doi:10.1016 / S0140-6736 (14) 61682-2. PMC 4340604. PMID 25530442.
- ^ Weatherall, D. J. (2015). „Thalassemias: Disorders of Globin Synthesis“. Williamsova hematologie (9e ed.). McGraw Hill Professional. p. 725. ISBN 9780071833011.
- ^ Cianciulli P (říjen 2008). "Léčba přetížení železem při talasemii". Pediatr Endocrinol Rev. 6 (Suppl 1): 208–13. PMID 19337180.
- ^ "Talasémie - příznaky a příčiny". Archivováno z původního dne 20. listopadu 2016. Citováno 4. dubna 2017.
- ^ Vogiatzi, Maria G; Macklin, Eric A; Fung, Ellen B; Cheung, Angela M; Vichinsky, Elliot; Olivieri, Nancy; Kirby, Melanie; Kwiatkowski, Janet L; Cunningham, Melody; Holm, Ingrid A; Lane, Joseph; Schneider, Robert; Fleisher, Martin; Grady, Robert W; Peterson, Charles C; Giardina, Patricia J (březen 2009). „Onemocnění kostí při talasemii: častý a stále nevyřešený problém“. Journal of Bone and Mineral Research. 24 (3): 543–557. doi:10,1359 / jbmr.080505. ISSN 0884-0431. PMC 3276604. PMID 18505376.
- ^ "Příznaky a příčiny - zvětšená slezina (splenomegalie) - Mayo Clinic". www.mayoclinic.org. Archivováno z původního dne 19. listopadu 2016. Citováno 2. února 2017.
- ^ Soliman, Ashraf T; Kalra, Sanjay; De Sanctis, Vincenzo (1. listopadu 2014). "Anémie a růst". Indian Journal of Endocrinology and Metabolism. 18 (7): S1–5. doi:10.4103/2230-8210.145038. PMC 4266864. PMID 25538873.
- ^ "Komplikace talasemie". Talasémie. Otevřete publikování. Archivováno z původního dne 3. října 2011. Citováno 27. září 2011.
- ^ Wambua S; Mwangi, Tabitha W .; Kortok, Mojžíš; Uyoga, Sophie M .; Macharia, Alex W .; Mwacharo, Jedidah K .; Weatherall, David J .; Snow, Robert W .; Marsh, Kevin; Williams, Thomas N. (květen 2006). „Vliv α + -thalasémie na výskyt malárie a jiných nemocí u dětí žijících na pobřeží Keni“. PLOS Medicine. 3 (5): e158. doi:10.1371 / journal.pmed.0030158. PMC 1435778. PMID 16605300.
- ^ Tassiopoulos S; Deftereos, Spyros; Konstantopoulos, Kostas; Farmakis, Dimitris; Tsironi, Maria; Kyriakidis, Michalis; Aessopos, Athanassios (2005). „Poskytuje heterozygotní beta-talasémie ochranu před onemocněním věnčitých tepen?“. Annals of the New York Academy of Sciences. 1054: 467–70. doi:10.1196 / annals.1345.068. PMID 16339699.
- ^ Robbins Basic Pathology, strana č: 428
- ^ Online Mendelian Inheritance in Man (OMIM): Hemoglobin - alfa lokus 1; HBA1 - 141800
- ^ Online Mendelian Inheritance in Man (OMIM): Hemoglobin - alfa lokus 2; HBA2 - 141850
- ^ A b Galanello, Renzo; Cao, Antonio (5. ledna 2011). "Alfa-talasémie". Genetika v medicíně. 13 (2): 83–88. doi:10.1097 / GIM.0b013e3181fcb468. ISSN 1098-3600. PMID 21381239.
- ^ A b "Základy anémie". WebMD. Citováno 9. května 2019.
- ^ Online Mendelian Inheritance in Man (OMIM): Hemoglobin - Beta Locus; HBB - 141900
- ^ Torres Lde S (březen 2015). "Hemoglobin D-Punjab: původ, distribuce a laboratorní diagnostika". Revista Brasileira de Hematologia e Hemoterapia. doi:10.1016 / j.bjhh.2015.02.007. PMID 25818823. Citováno 16. října 2020. Citovat deník vyžaduje
| deník =(Pomoc) - ^ „Jak jsou diagnostikovány thalassemie? - NHLBI, NIH“. www.nhlbi.nih.gov. Archivováno z původního dne 28. července 2017. Citováno 6. září 2017.
- ^ https://play.google.com/store/apps/details?id=com.gmail.sarahtinmaswala.challenge
- ^ „Screening nosičů ve věku genomické medicíny - ACOG“. www.acog.org. Archivováno z původního dne 25. února 2017. Citováno 24. února 2017.
- ^ Leung TN; Lau TK; Chung TKh (duben 2005). "Screening talasémie v těhotenství". Aktuální názor v porodnictví a gynekologii. 17 (2): 129–34. doi:10.1097 / 01.gco.0000162180.22984.a3. PMID 15758603. S2CID 41877258.
- ^ Loukopoulos, D (říjen 2011). „Hemoglobinopatie v Řecku: preventivní program za posledních 35 let“. Indický žurnál lékařského výzkumu. 134: 572–6. PMC 3237258. PMID 22089622.
- ^ Samavat A, Modell B (listopad 2004). „Íránský národní screeningový program na talasemii“. BMJ (Clinical Research Ed.). 329 (7475): 1134–7. doi:10.1136 / bmj.329.7475.1134. PMC 527686. PMID 15539666.
- ^ Petrou, Mary (1. ledna 2010). „Screening for beta thalassemia“. Indian Journal of Human Genetics. 16 (1): 1–5. doi:10.4103/0971-6866.64934. PMC 2927788. PMID 20838484.[trvalý mrtvý odkaz ]
- ^ A b Léčba pediatrické talasemie na eMedicína
- ^ Burdick CO; Ntaios, G .; Rathod, D. (březen 2009). „Oddělení znaku talasemie a nedostatku železa jednoduchou kontrolou“. Dopoledne. J. Clin. Pathol. 131 (3): 444, odpověď autora 445. doi:10.1309 / AJCPC09VRAXEASMH. PMID 19228649. Archivovány od originál dne 22. září 2014.
- ^ Harrisonovy principy interního lékařství (17. vydání). McGraw-Hill lékařský. Září 2008. str. 776. ISBN 978-0-07-164114-2.
- ^ Ngim, CF; Lai, NM; Hong, JY; Tan, SL; Ramadas, A; Muthukumarasamy, P; Thong, MK (28. května 2020). "Terapie růstovým hormonem pro lidi s talasemií". Cochrane Database of Systematic Reviews. 5: CD012284. doi:10.1002 / 14651858.CD012284.pub3. PMID 32463488.
- ^ A b C d E F G Neufeld, EJ (2010). "Aktualizace chelatátorů železa v talasémii". Hematologie. 2010: 451–5. doi:10.1182 / asheducation-2010.1.451. PMID 21239834.
- ^ "Ferriprox". Drugs.com. Americká společnost farmaceutů zdravotnického systému. Archivováno z původního dne 20. září 2016. Citováno 5. září 2016.
- ^ Kye Mon Min Swe (2013). "Zinkové doplňky pro léčbu talasemie a srpkovitých onemocnění". Cochrane Database of Systematic Reviews (6): CD009415. doi:10.1002 / 14651858.CD009415.pub2. PMID 23807756.
- ^ A b Gaziev, J; Lucarelli, G (červen 2011). "Transplantace hematopoetických kmenových buněk pro talasemii". Současný výzkum a terapie kmenových buněk. 6 (2): 162–9. doi:10.2174/157488811795495413. PMID 21190532.
- ^ Sabloff, M; Chandy, M; Wang, Z; Logan, BR; Ghavamzadeh, A; Li, CK; Irfan, SM; Bredeson, CN; et al. (2011). „Transplantace kostní dřeně shodné s HLA pro β-thalassemia major“. Krev. 117 (5): 1745–50. doi:10.1182 / krev-2010-09-306829. PMC 3056598. PMID 21119108.
- ^ Jagannath, Vanitha A .; Fedorowicz, Zbys; Al Hajeri, Amani; Sharma, Akshay (30. listopadu 2016). „Transplantace hematopoetických kmenových buněk u lidí s ß-thalassemia major“. Cochrane Database of Systematic Reviews. 11: CD008708. doi:10.1002 / 14651858.CD008708.pub4. ISSN 1469-493X. PMC 6492419. PMID 27900772.
- ^ Fisher, Sheila A; Cutler, Antony; Doree, Carolyn; Brunskill, Susan J; Stanworth, Simon J; Navarrete, Cristina; Girdlestone, John (30. ledna 2019). Cochrane Haematological Malignancies Group (ed.). „Mesenchymální stromální buňky jako léčba nebo profylaxe pro akutní nebo chronické onemocnění štěp proti hostiteli u příjemců transplantace hematopoetických kmenových buněk (HSCT) s hematologickým stavem“. Cochrane Database of Systematic Reviews. 1: CD009768. doi:10.1002 / 14651858.CD009768.pub2. PMC 6353308. PMID 30697701.
- ^ Sodani, P; Isgrò, A; Gaziev, J; Paciaroni, K; Marziali, M; Simone, MD; Roveda, A; De Angelis, G; et al. (2011). „Transplantace hla-haploidentických kmenových buněk s deplecí T buněk u mladých pacientů s talasemií“. Pediatrické zprávy. 3 (Suppl 2): e13. doi:10.4081 / pr.2011.s2.e13. PMC 3206538. PMID 22053275.
- ^ A b John P. Greer JP, Arber DA, Glader B, et al. Wintrobe's Clinical Hematology 2013. ISBN 9781451172683
- ^ Waheed, Fazeela; Fishter, Colleen; Awofeso, AwoNiyi; Stanley, David (červenec 2016). „Screening nosičů na beta-talasémii na Maledivách: vnímání rodičů postižených dětí, kteří se screeningu nezúčastnili, a jeho důsledky“. Journal of Community Genetics. 7 (3): 243–253. doi:10.1007 / s12687-016-0273-5. PMC 4960032. PMID 27393346.
- ^ Modiano, G .; Morpurgo, G; Terrenato, L; Novelletto, A; Di Rienzo, A; Colombo, B; Purpura, M; Mariani, M; et al. (1991). „Ochrana před morbiditou na malárii: Blízká fixace genu α-thalasémie u nepálské populace“. American Journal of Human Genetics. 48 (2): 390–7. PMC 1683029. PMID 1990845.
- ^ Terrenato, L; Shrestha, S; Dixit, KA; Luzzatto, L; Modiano, G; Morpurgo, G; Arese, P (únor 1988). „Snížená nemocnost na malárii u Tharuů ve srovnání se sympatickými populacemi v Nepálu“. Annals of Tropical Medicine and Parasitology. 82 (1): 1–11. doi:10.1080/00034983.1988.11812202. PMID 3041928.
- ^ E. Goljan, Pathology, 2. vyd. Mosby Elsevier, série rychlých recenzí.[stránka potřebná ]
- ^ "Thalassemia" (v thajštině). Ústav lékařských věd. Září 2011. Archivovány od originál dne 25. září 2011.
- ^ Galanello, Renzo; Origa, Raffaella (2010). „Beta-talasémie“. Orphanet Journal of Rare Diseases. 5 (1): 11. doi:10.1186/1750-1172-5-11. PMC 2893117. PMID 20492708.
- ^ Galanello, Renzo; Origa, Raffaella (2010). „Beta-talasémie“. Orphanet Journal of Rare Diseases. 5 (1): 11. doi:10.1186/1750-1172-5-11. PMC 2893117. PMID 20492708.
- ^ Vichinsky, Elliott P. (1. listopadu 2005). "Měnící se vzorce talasemie po celém světě". Annals of the New York Academy of Sciences. 1054 (1): 18–24. Bibcode:2005NYASA1054 ... 18V. doi:10.1196 / annals.1345.003. ISSN 1749-6632. PMID 16339647.
- ^ A b POČASÍ, DAVID J. (listopad 2005). „Hlavní projev: Výzva talasémie pro rozvojové země“. Annals of the New York Academy of Sciences. 1054 (1): 11–17. Bibcode:2005NYASA1054 ... 11W. doi:10.1196 / annals.1345.002. PMID 16339646.
- ^ θάλασσα. Liddell, Henry George; Scott, Robert; Řecko-anglický lexikon na Projekt Perseus.
- ^ αἷμα v Liddell a Scott.
- ^ Whipple GH, Bradford WI. Am J Dis Child 1932; 44: 336
- ^ „Spanish Baby Engineered to Cure Brother“. Archivovány od originál dne 15. října 2008.
- ^ Chovatel jeho sestry: Krev bratra je požehnáním života Archivováno 22. září 2009 v Wayback Machine, Časy Indie, 17. září 2009
- ^ A b Negre, Olivier; Eggimann, Anne-Virginie; Beuzard, Yves; Ribeil, Jean-Antoine; Bourget, Philippe; Borwornpinyo, Suparerk; Hongeng, Suradej; Hacein-Bey, Salima; Cavazzana, Marina; Leboulch, Philippe; Payen, Emmanuel (únor 2016). "Genová terapie β-hemoglobinopatií pomocí lentivirového přenosu β - genu". Lidská genová terapie. 27 (2): 148–165. doi:10.1089 / hum.2016.007. PMC 4779296. PMID 26886832.
- ^ Biffi, A (19. dubna 2018). „Genová terapie jako léčebná alternativa pro β-talasemii“. The New England Journal of Medicine. 378 (16): 1551–1552. doi:10.1056 / NEJMe1802169. PMID 29669229.
- ^ Lidonnici, MR; Ferrari, G (květen 2018). "Genová terapie a strategie úpravy genů pro hemoglobinopatie". Krevní buňky, molekuly a nemoci. 70: 87–101. doi:10.1016 / j.bcmd.2017.12.001. PMID 29336892.
- ^ A b Wienert, B; Martyn, GE; Funnell, APW; Quinlan, KGR; Crossley, M (1. října 2018). „Wake-up Sleepy Gene: Reactivating Fetal Globin for β-Hemoglobinopathies“. Trendy v genetice. 34 (12): 927–940. doi:10.1016 / j.tig.2018.09.004. PMID 30287096.
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |
- Talasémie na Curlie
- Učení o talasemii publikoval Národní institut pro výzkum lidského genomu.