MECP2 - MECP2






MECP2 (protein vázající methyl CpG 2) je gen[5] který kóduje protein MECP2.[6] MECP2 se jeví jako nezbytný pro normální funkci nervové buňky. Protein se zdá být zvláště důležitý pro zralé nervové buňky, kde je přítomen ve vysokých hladinách. Je pravděpodobné, že protein MECP2 se bude podílet na vypnutí („potlačování“ nebo „umlčení ") několik dalších genů. To brání genům v tvorbě proteinů, pokud nejsou potřebné. Nedávné práce ukázaly, že MECP2 může také aktivovat další geny.[7] Gen MECP2 se nachází na dlouhém (q) rameni X chromozom v pásmu 28 ("Xq28") od základní pár 152 808 110 na základní pár 152 878 611.
MECP2 je důležitým čtenářem methylace DNA. Jeho doména vázající methyl-CpG (MBD) rozpoznává a váže se 5-mC regionech. MECP2 je vázán na X a podléhá X deaktivace. Genové mutace MECP2 jsou příčinou většiny případů Rettův syndrom, progresivní neurologická vývojová porucha a jedna z nejčastějších příčin kognitivního postižení u žen.[8]
Funkce
MECP2 protein se vyskytuje ve všech buňky v těle, včetně mozek, působící jako transkripční represor a aktivátor, v závislosti na kontextu. Myšlenka, že MECP2 funguje jako aktivátor, je však relativně nová a zůstává kontroverzní.[9] V mozku se nachází ve vysokých koncentracích v neurony a je spojena se zráním centrální nervový systém (CNS) a při formování synaptické kontakty.[10]
Mechanismus účinku
Protein MeCP2 se váže na formy DNA které byly methylovaný. Protein MeCP2 pak interaguje s jinými proteiny za vzniku komplexu, který vypíná gen. MeCP2 dává přednost vazbě na místa v genomu chemickou změnou provedenou na a cytosin (C) když se vyskytuje v konkrétní sekvenci DNA, "CpG ". Toto je forma Methylace DNA. Mnoho genů má CpG ostrovy, které se často vyskytují blízko začátku genu. MECP2 se na tyto ostrovy ve většině případů neváže, protože nejsou methylované. The výraz několik genů může být regulováno methylací jejich CpG ostrova a MECP2 může hrát roli v jejich podmnožině. Vědci dosud neurčili, na které geny se zaměřuje protein MeCP2, ale tyto geny jsou pravděpodobně důležité pro normální funkci centrálního nervového systému. První rozsáhlé mapování vazebných míst MECP2 v neuronech však zjistilo, že pouze 6% vazebných míst je na ostrovech CpG a že 63% promotorů vázaných na MECP2 je aktivně exprimováno a pouze 6% je vysoce methylováno, což naznačuje, že Hlavní funkcí MECP2 je něco jiného než umlčení methylovaných promotorů.[11]
Po navázání MeCP2 kondenzuje chromatin struktura, tvoří komplex s histonové deacetylázy (HDAC) nebo blokovat transkripční faktory přímo. Novější studie prokázaly, že MeCP2 může také fungovat jako transkripční aktivátor prostřednictvím náboru transkripčního faktoru CREB1. Toto bylo neočekávané zjištění, které naznačuje, že MeCP2 je klíčový regulátor transkripce s potenciálně dvojí rolí v genové expresi. Ve skutečnosti se zdá, že většina genů regulovaných MeCP2 je spíše aktivována než potlačována.[12] Zůstává však kontroverzní, zda MeCP2 reguluje tyto geny přímo, nebo zda jsou tyto změny sekundární povahy.[9] Další studie ukázaly, že MeCP2 se v některých případech může vázat přímo na nemethylovanou DNA.[13] MeCP2 se podílí na regulaci otiskovaných genů a lokusů, které zahrnují UBE3A a DLX5.[14]
Snížená exprese MECP2 v Mecp2 +/- nervové kmenové buňky způsobí zvýšení stárnutí, narušení proliferační kapacity a akumulace neopravených Poškození DNA.[15] Po ošetření buněk Mecp2 +/- jedním ze tří různých činidel poškozujících DNA se buňky hromadily více poškození DNA a byly náchylnější k buněčné smrti než kontrolní buňky.[15] Byl učiněn závěr, že snížená exprese MECP2 způsobuje sníženou kapacitu na opravit DNA a to pravděpodobně přispívá k neurologickému úpadku.[15]
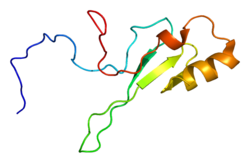
Struktura
MECP2 je součástí rodiny proteinů vázajících methyl-CpG (MBD), ale má své vlastní jedinečné rozdíly, které ho odlišují od skupiny. Má dvě funkční domény:
- methyl-cytosin -binding doména (MBD) složená z 85 aminokyseliny; a
- transkripční represivní doména (TRD) složená ze 104 aminokyselin
Doména MBD tvoří klín a připojuje se k methylovaným CpG místům na vláknech DNA. Oblast TRD poté reaguje se SIN3A a získává histonové deacetylázy (HDAC).[16] Na karboxylovém konci se také nacházejí neobvyklé, opakující se sekvence. Tato oblast úzce souvisí s rodinou vidlicových hlav na úrovni aminokyselin.[17]
Role v nemoci
Role MECP2 u onemocnění je primárně spojena buď se ztrátou funkce (pod expresí) genu MECP2, jako u Rettův syndrom nebo v zisku funkce (nad výrazem) jako v Syndrom duplikace MECP2. Mnoho mutací bylo spojeno se ztrátou exprese genu MECP2 a byly identifikovány u pacientů s Rettovým syndromem. Tyto mutace zahrnují změny v jediné DNA základní páry (SNP ), inzerce nebo delece DNA v MECP2 gen a změny, které ovlivňují způsob, jakým jsou informace o genu zpracovány na protein (Sestřih RNA ). Mutace v genu mění strukturu proteinu MeCP2 nebo vedou ke sníženému množství proteinu. Výsledkem je, že se protein nedokáže vázat na DNA nebo zapnout nebo vypnout jiné geny. Geny, které jsou normálně potlačovány MeCP2, zůstávají aktivní, pokud jejich produkty nejsou potřeba. Jiné geny, které jsou normálně aktivovány MeCP2, zůstávají neaktivní, což vede k nedostatku genového produktu. Tato vada pravděpodobně narušuje normální fungování nervových buněk, což vede ke známkám a příznakům Rettova syndromu.
Rettův syndrom se vyskytuje hlavně u dívek s a prevalence přibližně 1 z každých 10 000. Pacienti se rodí s velmi těžkými známkami poruchy, ale přibližně po šesti měsících až roce a půl se začnou snižovat řečové a motorické funkce. Poté následuje záchvaty, zpomalení růstu a kognitivní a motorické poškození.[18] Lokalita MECP2 je X-vázaný a alely způsobující onemocnění jsou dominantní. Vzhledem ke své prevalenci u žen je spojován s mužskou letalitou nebo s převládajícím přenosem otcovským chromozomem X; ve vzácných případech však může být Rettovým syndromem postižen i některý muž.[19] Muži s genovou duplikací MECP-2 na lokusu Xq28 jsou také v dětství ohroženi opakovanými infekcemi a meningitidou.
Mutace v genu MECP2 byly také identifikovány u lidí s několika dalšími poruchami ovlivňujícími centrální nervový systém. Například mutace MECP2 jsou spojeny s některými případy středně těžké až těžké mentální retardace spojené s X. Mutace v genu byly také nalezeny u mužů s těžkou mozkovou dysfunkcí (neonatální encefalopatie ), kteří žijí jen do raného dětství. Kromě toho několik lidí s rysy Rettova syndromu a Angelmanov syndrom (stav charakterizovaný mentální retardací, problémy s pohybem a nevhodným smíchem a vzrušivostí) mají mutace v genu MECP2. A konečně, v některých případech byly hlášeny mutace MECP2 nebo změny v aktivitě genu autismus (vývojová porucha, která ovlivňuje komunikaci a sociální interakci).[20]
Novější studie uváděly genetické polymorfismy v genech MeCP2 u pacientů s systémový lupus erythematodes (SLE).[21] SLE je systémové autoimunitní onemocnění, které může postihnout více orgánů. Polymorfismy MeCP2 byly dosud hlášeny u pacientů s evropským a asijským lupusem.
Genetická ztráta MECP2 byla identifikována jako změna vlastností buněk v locus ceruleus výhradní zdroj noradrenergní inervace do mozková kůra a hipokampus.[22]
Vědci dospěli k závěru, že „Protože tyto neurony jsou stěžejním zdrojem norepinefrinu v celém mozkovém kmeni a předním mozku a podílejí se na regulaci různých funkcí narušených u Rettova syndromu, jako je dýchání a poznávání, předpokládáme, že lokus ceruleus je kritickým místem na která ztráta MECP2 vede k dysfunkci CNS. “[22]
Interaktivní mapa cest
Kliknutím na geny, proteiny a metabolity níže můžete navštívit související články. [§ 1]

Interakce
MECP2 bylo prokázáno komunikovat s SKI protein[23] a Korepresor jaderného receptoru 1.[23] V neuronálních buňkách se předpokládá, že mRNA MECP2 interaguje miR-132, který umlčí expresi proteinu. To tvoří součást homeostatického mechanismu, který by mohl regulovat hladiny MECP2 v mozku.[24]
MeCP2 a hormony
MeCP2 ve vyvíjejícím se potkaním mozku reguluje důležitý sociální vývoj sexuálně dimorfně. Hladiny MeCP2 se liší u mužů a žen ve vyvíjejícím se potkaním mozku 24 hodin po narození amygdala a hypotalamus, ale tento rozdíl již není pozorován 10 dní po narození. Konkrétně muži vyjadřují méně MeCP2 než ženy,[25] a to odpovídá časovému období citlivému na steroidy v mozku krys novorozence. Snížení MeCP2 s Malá interferující RNA (siRNA) během prvních několika dnů života snižují mužské úrovně juvenilního sociálního herního chování na typické ženské úrovně, neovlivňují však herní chování mladistvých žen.[26]
MeCP2 je důležitý při organizování hormonálního chování a pohlavních rozdílů ve vyvíjející se krysí amygdale. Zdá se, že MeCP2 reguluje arginin vazopresin (AVP) a androgenní receptor (AR) produkce u samců potkanů, ale ne u samic. Je známo, že vazopresin reguluje mnoho společenských chování, včetně párových vazeb[27] a sociální uznání.[28] Zatímco krysy samců mají obvykle vyšší hladinu vazopresinu v amygdale,[29] Snížení MeCP2 během prvních 3 dnů života způsobuje trvalé snížení vazopresinu na ženské typické úrovně v této oblasti mozku, které přetrvávalo až do dospělosti. Samci potkanů se sníženými hladinami MeCP2 také vykazují významné snížení AR dva týdny po infuzi, ale tento účinek zmizel v dospělosti.[30]
Stres v raném životě
MeCP2 sleduje reakci na stres v raném věku. Stres raného života koreluje s hyperfosforylací proteinu MeCP2 v paraventrikulární jádro hypotalamu.[31] To tedy způsobuje snížené obsazení MeCP2 v promotorové oblasti genu AVP, a proto zvýšené hladiny AVP. Vasopresin je primární hormon podílející se na ose Hypothalmic-hypofýza-nadledvina, propojení v mozku, které reguluje zpracování a reakci na stres. Snížené fungování proteinu MeCP2 tak zvyšuje regulaci nervové stresové reakce.
Reference
- ^ A b C GRCh38: Vydání souboru 89: ENSG00000169057 - Ensembl, Květen 2017
- ^ A b C GRCm38: Vydání souboru 89: ENSMUSG00000031393 - Ensembl, Květen 2017
- ^ „Human PubMed Reference:“. Národní centrum pro biotechnologické informace, Americká národní lékařská knihovna.
- ^ „Myš PubMed Reference:“. Národní centrum pro biotechnologické informace, Americká národní lékařská knihovna.
- ^ Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (Říjen 1999). „Rettův syndrom je způsoben mutacemi v XEC vázaném MECP2 kódujícím protein vázající methyl-CpG 2“. Nat. Genet. 23 (2): 185–8. doi:10.1038/13810. PMID 10508514. S2CID 3350350.
- ^ Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A (červen 1992). "Čištění, sekvence a buněčná lokalizace nového chromozomálního proteinu, který se váže na methylovanou DNA". Buňka. 69 (6): 905–14. doi:10.1016 / 0092-8674 (92) 90610-O. PMID 1606614. S2CID 6825994.
- ^ Chahrour M a kol. (2008). „MECP2, klíčový přispěvatel k neurologickému onemocnění, aktivuje a potlačuje transkripci“. Věda. 320 (5880): 1224–9. doi:10.1126 / science.1153252. PMC 2443785. PMID 18511691.
- ^ „Entrez Gene: MECP2 methyl CpG binding protein 2 (Rettův syndrom)“.
- ^ A b Cohen S, Zhou Z, Greenberg ME (květen 2008). „Medicína. Aktivace represoru“. Věda. 320 (5880): 1172–3. doi:10.1126 / science.1159146. PMC 2857976. PMID 18511680.
- ^ Luikenhuis S, Giacometti E, Beard CF, Jaenisch R (duben 2004). "Exprese MeCP2 v postmitotických neuronech zachraňuje Rettův syndrom u myší". Proc. Natl. Acad. Sci. USA. 101 (16): 6033–8. doi:10.1073 / pnas.0401626101. PMC 395918. PMID 15069197.
- ^ Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM (prosinec 2007). „Integrované epigenomické analýzy neuronového MeCP2 odhalují roli interakce s aktivními geny na velké vzdálenosti“. Proc. Natl. Acad. Sci. USA. 104 (49): 19416–21. doi:10.1073 / pnas.0707442104. PMC 2148304. PMID 18042715.
- ^ Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY (květen 2008). „MeCP2, klíčový přispěvatel k neurologickému onemocnění, aktivuje a potlačuje transkripci“. Věda. 320 (5880): 1224–9. doi:10.1126 / science.1153252. PMC 2443785. PMID 18511691.
- ^ Georgel PT, Horowitz-Scherer RA, Adkins N, Woodcock CL, Wade PA, Hansen JC (srpen 2003). "Chromatinové zhutnění lidským MeCP2. Sestavení nových sekundárních chromatinových struktur bez přítomnosti methylace DNA". J. Biol. Chem. 278 (34): 32181–8. doi:10,1074 / jbc.M305308200. PMID 12788925.
- ^ LaSalle JM (2007). „Odysea MeCP2 a otisk rodičů“. Epigenetika. 2 (1): 5–10. doi:10.4161 / epi.2.1.3697. PMC 1866173. PMID 17486180.
- ^ A b C Alessio N, Riccitiello F, Squillaro T, Capasso S, Del Gaudio S, Di Bernardo G, Cipollaro M, Melone MAB, Peluso G, Galderisi U (březen 2018). „Neurální kmenové buňky z myšího modelu Rettova syndromu jsou náchylné k stárnutí, vykazují sníženou schopnost vyrovnat se s genotoxickým stresem a jsou narušeny v procesu diferenciace.“. Exp. Mol. Med. 50 (3): 1. doi:10.1038 / s12276-017-0005-x. PMC 6118406. PMID 29563495.
- ^ Wakefield RI, Smith BO, Nan X, Free A, Soteriou A, Uhrin D, Bird AP, Barlow PN (září 1999). "Struktura řešení domény z MeCP2, která se váže na methylovanou DNA". J. Mol. Biol. 291 (5): 1055–65. doi:10.1006 / jmbi.1999.3023. PMID 10518942.
- ^ Paul A. Wade (prosinec 2001). "Proteiny vázající methyl CpG a transkripční represi" (PDF). BioEssays. 23 (12): 1131–1137. doi:10.1002 / bies.10008. PMID 11746232. S2CID 37525856. Archivovány od originál (PDF) dne 2007-08-14.
- ^ Caballero IM, Hendrich B (duben 2005). „MeCP2 v neuronech: blížící se příčinám Rettova syndromu“. Hučení. Mol. Genet. 14 Specifikace č. 1: R19–26. doi:10,1093 / hmg / ddi102. PMID 15809268.
- ^ Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM (březen 2004). „Několik cest reguluje expresi MeCP2 v normálním vývoji mozku a vykazuje vady poruch autistického spektra“. Hučení. Mol. Genet. 13 (6): 629–39. doi:10,1093 / hmg / ddh063. PMID 14734626.
- ^ Hunt, Katie (12. ledna 2016). „Čínští vědci vytvářejí opice s genem autismu“. Zprávy CNN. Citováno 2016-01-27.
- ^ Sawalha AH, Webb R, Han S, Kelly JA, Kaufman KM, Kimberly RP, Alarcón-Riquelme ME, James JA, Vyse TJ, Gilkeson GS, Choi CB, Scofield RH, Bae SC, Nath SK, Harley JB (2008). Jin D (ed.). „Běžné varianty v rámci MECP2 představují riziko systémového lupus erythematodes“. PLOS ONE. 3 (3): e1727. doi:10.1371 / journal.pone.0001727. PMC 2253825. PMID 18320046.

- ^ A b Taneja P, Ogier M, Brooks-Harris G, Schmid DA, Katz DM, Nelson SB (2009). „Patofyziologie neuronů Locus Ceruleus u myšího modelu Rettova syndromu“. Journal of Neuroscience. 29 (39): 12187–12195. doi:10.1523 / JNEUROSCI.3156-09.2009. PMC 2846656. PMID 19793977.
- ^ A b Kokura K, Kaul SC, Wadhwa R, Nomura T, Khan MM, Shinagawa T, Yasukawa T, Colmenares C, Ishii S (září 2001). „Rodina proteinů Ski je vyžadována pro transkripční represi zprostředkovanou MeCP2“. J. Biol. Chem. 276 (36): 34115–21. doi:10,1074 / jbc.M105747200. PMID 11441023.
- ^ Klein ME, Lioy DT, Ma L, Impey S, Mandel G, Goodman RH (prosinec 2007). „Homeostatická regulace exprese MeCP2 mikroRNA indukovanou CREB“. Nat. Neurosci. 10 (12): 1513–4. doi:10.1038 / nn2010. PMID 17994015. S2CID 29308441.
- ^ Kurian JR, Forbes-Lorman RM, Auger AP (září 2007). „Rozdíl pohlaví v expresi mecp2 během kritického období vývoje mozku potkana“. Epigenetika. 2 (3): 173–8. doi:10.4161 / epi.2.3.4841. PMID 17965589.
- ^ Kurian JR, Bychowski ME, Forbes-Lorman RM, Auger CJ, Auger AP (červenec 2008). „Mecp2 organizuje mladistvé sociální chování způsobem specifickým pro pohlaví“. J. Neurosci. 28 (28): 7137–42. doi:10.1523 / JNEUROSCI.1345-08.2008. PMC 2569867. PMID 18614683.
- ^ Winslow JT, Hastings N, Carter CS, Harbaugh CR, Insel TR (říjen 1993). „Role centrálního vazopresinu při párování v monogamních prérijních hraboších“. Příroda. 365 (6446): 545–8. doi:10.1038 / 365545a0. PMID 8413608. S2CID 4333114.
- ^ Bielsky IF, Hu SB, Szegda KL, Westphal H, Young LJ (březen 2004). „Hluboké zhoršení sociálního uznání a snížení úzkostného chování u myší s knockoutem receptoru vazopresinu V1a“. Neuropsychofarmakologie. 29 (3): 483–93. doi:10.1038 / sj.npp.1300360. PMID 14647484.
- ^ De Vries GJ, Panzica GC (2006). „Sexuální diferenciace centrálních vazopresinových a vasotocinových systémů u obratlovců: různé mechanismy, podobné koncové body“. Neurovědy. 138 (3): 947–55. doi:10.1016 / j.neuroscience.2005.07.050. PMC 1457099. PMID 16310321.
- ^ Forbes-Lorman RM, Rautio JJ, Kurian JR, Auger AP, Auger CJ (březen 2012). „Novorozenecký MeCP2 je důležitý pro organizaci pohlavních rozdílů ve expresi vazopresinu“. Epigenetika. 7 (3): 230–8. doi:10.4161 / epi.7.3.19265. PMC 3335947. PMID 22430799.
- ^ Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmühl Y, Fischer D, Holsboer F, Wotjak CT, Almeida OF, Spengler D (prosinec 2009). „Programy dynamické methylace DNA přetrvávající nepříznivé účinky stresu v raném věku“. Nat. Neurosci. 12 (12): 1559–66. doi:10.1038 / č. 2436. PMID 19898468. S2CID 3328884.
Další čtení
- Chahrour M, Zoghbi HY (2007). „Příběh Rettova syndromu: od kliniky k neurobiologii“. Neuron. 56 (3): 422–37. doi:10.1016 / j.neuron.2007.10.001. PMID 17988628. S2CID 16266882.
- Carney RM, Wolpert CM, Ravan SA, Shahbazian M, Ashley-Koch A, Cuccaro ML, Vance JM, Pericak-Vance MA (2003). "Identifikace mutací MeCP2 u řady žen s autistickou poruchou". Pediatr Neurol. 28 (3): 205–11. doi:10.1016 / S0887-8994 (02) 00624-0. PMID 12770674.
- Kerr AM, Ravine D (2003). „Recenze: průlom s Rettovým syndromem“. J Intellect Disabil Res. 47 (Pt 8): 580–7. doi:10.1046 / j.1365-2788.2003.00506.x. PMID 14641805.
- Neul JL, Zoghbi HY (2004). „Rettův syndrom: prototypová neurovývojová porucha“. Neuro vědec. 10 (2): 118–28. doi:10.1177/1073858403260995. PMID 15070486. S2CID 9617631.
- Schanen C, Houwink EJ, Dorrani N, Lane J, Everett R, Feng A, Cantor RM, Percy A (2004). "Fenotypové projevy mutací MECP2 u klasického a atypického Rettova syndromu". Am J Med Genet A. 126 (2): 129–40. doi:10,1002 / ajmg.a.20571. PMID 15057977. S2CID 32897044.
- Van den Veyver IB, Zoghbi HY (2001). „Mutace v genu kódujícím protein vázající methyl-CpG 2 způsobují Rettův syndrom“. Brain Dev. 23 (Suppl 1): S147–51. doi:10.1016 / S0387-7604 (01) 00376-X. PMID 11738862. S2CID 26138178.
- Webb T, Latif F (2001). "Rettův syndrom a gen MECP2". J Med Genet. 38 (4): 217–23. doi:10,1136 / jmg.38.4.217. PMC 1734858. PMID 11283201.
- Shahbazian MD, Zoghbi HY (2003). „Rettův syndrom a MeCP2: propojení epigenetiky a neuronální funkce“. Dopoledne. J. Hum. Genet. 71 (6): 1259–72. doi:10.1086/345360. PMC 378559. PMID 12442230.
- Moog U, Smeets EE, van Roozendaal KE a kol. (2003). "Neurodevelopmentální poruchy u mužů související s genem způsobujícím Rettův syndrom u žen (MECP2)". Eur. J. Pediatr. Neurol. 7 (1): 5–12. doi:10.1016 / S1090-3798 (02) 00134-4. PMID 12615169.
- Miltenberger-Miltenyi G, Laccone F (2004). "Mutace a polymorfismy v lidském methylovém CpG-vázajícím proteinu MECP2". Hučení. Mutat. 22 (2): 107–15. doi:10,1002 / humu.10243. PMID 12872250.
- Tkaní LS, Ellaway CJ, Gécz J, Christodoulou J (2006). „Rettův syndrom: klinický přehled a genetická aktualizace“. J. Med. Genet. 42 (1): 1–7. doi:10.1136 / jmg.2004.027730. PMC 1735910. PMID 15635068.
- Bapat S, Galande S (2005). „Asociace vinou: identifikace DLX5 jako cíle pro MeCP2 poskytuje molekulární vazbu mezi genomovým otiskem a Rettovým syndromem“. BioEssays. 27 (7): 676–80. doi:10.1002 / bies.20266. PMID 15954098.
- Zlatanova J (2005). Msgstr "MeCP2: připojení chromatinu a dále". Biochem. Cell Biol. 83 (3): 251–62. doi:10.1139 / o05-048. PMID 15959553.
- Kaufmann WE, Johnston MV, Blue ME (2006). „Exprese a funkce MeCP2 během vývoje mozku: důsledky pro patogenezi a klinický vývoj Rettova syndromu“. Brain Dev. 27 (Suppl 1): S77 – S87. doi:10.1016 / j.braindev.2004.10.008. PMID 16182491. S2CID 702975.
- Armstrong DD (2006). „Můžeme spojit nedostatek MeCP2 se strukturálními a chemickými abnormalitami v mozku Retta?“. Brain Dev. 27 (Suppl 1): S72 – S76. doi:10.1016 / j.braindev.2004.10.009. PMID 16182497. S2CID 45587850.
- Santos M, Coelho PA, Maciel P (2006). "Remodelace chromatinu a neuronální funkce: vzrušující odkazy". Geny, mozek a chování. 5 (Suppl 2): 80–91. doi:10.1111 / j.1601-183X.2006.00227.x. PMID 16681803.
- Bienvenu T, Chelly J (2006). „Molekulární genetika Rettova syndromu: když není metylace DNA rozpoznána“. Genetika hodnocení přírody. 7 (6): 415–26. doi:10.1038 / nrg1878. PMID 16708070. S2CID 28215286.
- Francke U (2007). "Mechanismy nemoci: neurogenetika nedostatku MeCP2". Přírodní neurologie z klinické praxe. 2 (4): 212–21. doi:10.1038 / ncpneuro0148. PMID 16932552. S2CID 22710951.
externí odkazy
- International Rett Syndrome Foundation
- Podpora a výzkumná charita Rett UK
- Trust pro výzkum Rettova syndromu
- Ensembl Gen ref Protein ref
- GeneCard
- RettBASE: Databáze variant IRSA MECP2
- GeneReview / NIH / UW záznam o poruchách souvisejících s MECP2
- GeneReviews / NCBI / NIH / UW záznam o syndromu duplikace MECP2
- Britský web pro rodiny postižené MECP2.
- Web pro rodiny postižené MECP2.
- Oficiální stránky français sur la duplikace MeCP2
Galerie PDB | |
|---|---|
|