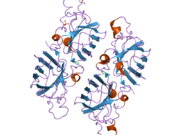SOD1 - SOD1

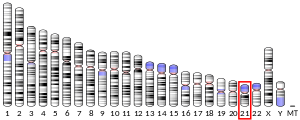




Superoxid dismutáza [Cu-Zn] také známý jako superoxiddismutáza 1 nebo SOD1 je enzym že u lidí je kódován SOD1 gen, umístěný na chromozom 21. SOD1 je jedním ze tří lidských superoxid dismutázy.[5][6] Je zapleten do apoptóza a rodinné Amyotrofní laterální skleróza.[6]
Struktura
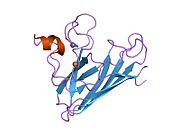
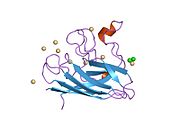
SOD1 je 32 kDa homodimer který tvoří β-hlaveň a obsahuje intramolekulární disulfidovou vazbu a dvoujaderné místo Cu / Zn v každé podjednotce. Toto místo Cu / Zn obsahuje ionty mědi a zinku a je zodpovědné za katalýzu nepřiměřenost z superoxid na peroxid vodíku a dioxygen.[7][8] Proces zrání tohoto proteinu je složitý a není zcela objasněn, zahrnuje selektivní vazbu iontů mědi a zinku, tvorbu intra-podjednotky disulfidová vazba mezi Cys-57 a Cys-146 a dimerizace obou podjednotek. Chaperon mědi pro Sod1 (CCS) usnadňuje vkládání mědi a oxidaci disulfidu. Ačkoli je SOD1 syntetizován v cytosolu a může tam zrát, musí být do intermembránového prostoru vložen zlomek exprimovaného a stále nezralého SOD1 zaměřeného na mitochondrie. Tam vytváří disulfidovou vazbu, i když ne metalační, potřebnou pro její zrání.[8] Zralý protein je vysoce stabilní,[9] ale nestabilní, když je ve formě bez obsahu kovů a disulfidu.[7][8][9] To se projevuje in vitro, protože ztráta kovových iontů vede ke zvýšené agregaci SOD1, a na modelech onemocnění, kde je u nerozpustného SOD1 pozorována nízká metalizace. Kromě toho by se povrchově exponované redukované cysteiny mohly účastnit disulfidu síťování a tedy agregace.[7]
Funkce
SOD1 váže ionty mědi a zinku a je jednou ze tří superoxiddismutáz odpovědných za ničení volných látek superoxid radikály v těle. Zakódováno isozym je rozpustný cytoplazmatický a mitochondriální mezimembránový prostorový protein, který působí jako homodimer pro přeměnu přirozeně se vyskytujících, ale škodlivých, superoxidových radikálů na molekulární kyslík a peroxid vodíku.[8][10] Peroxid vodíku pak může být rozložen jiným enzymem zvaným kataláza.
SOD1 byl postulován do lokalizovat do vnější mitochondriální membrána (OMM), kde by se generovaly superoxidové anionty, nebo mezimembránový prostor. Přesné mechanismy jeho lokalizace zůstávají neznámé, ale jeho agregace do OMM byla přičítána jeho asociaci s BCL-2. Divoký typ SOD1 prokázal antiapoptotické vlastnosti v nervových kulturách, zatímco u mutantního SOD1 bylo pozorováno, že podporuje apoptózu v mitochondriích míchy, ale ne u játra mitochondrie, i když je to u obou vyjádřeno stejně. Dva modely naznačují, že SOD1 inhibuje apoptózu interakcí s BCL-2 proteiny nebo samotné mitochondrie.[6]
Klinický význam
Role v oxidačním stresu
Nejpozoruhodnější je, že SOD1 je stěžejní reaktivní formy kyslíku (ROS) uvolňování během oxidačního stresu ischemicko-reperfuzním poškozením, konkrétně v myokardu jako součást infarkt (také známý jako ischemická choroba srdeční ). Ischemická choroba srdeční, která je výsledkem okluze jednoho z hlavních Koronární tepny, je v současné době stále hlavní příčinou nemocnost a úmrtnost v západní společnosti.[11][12] Během ischemické reperfúze uvolňování ROS podstatně přispívá k poškození a smrti buněk přímým účinkem na buňku i apoptotickými signály. Je známo, že SOD1 má schopnost omezovat škodlivé účinky ROS. Jako takový je SOD1 důležitý pro své kardioprotektivní účinky.[13] Kromě toho se SOD1 podílí na kardioprotekci proti ischemicko-reperfuznímu poškození, například během ischemická stabilizace srdce.[14] I když je známo, že velká dávka ROS vede k poškození buněk, mírné uvolňování ROS z mitochondrií, ke kterému dochází během krátkých epizod ischemie, může hrát významnou spouštěcí roli v drahách signální transdukce ischemické předběžné úpravy vedoucí ke snížení poškození buněk. Dokonce bylo pozorováno, že během tohoto uvolňování ROS hraje SOD1 důležitou roli při regulaci apoptotické signalizace a buněčné smrti.
V jedné studii byly delece v genu hlášeny ve dvou rodinných případech keratokonus.[15] Myši bez SOD1 zvýšily úbytek svalové hmoty související s věkem (sarkopenie ), časný vývoj šedý zákal, makulární degenerace, brzlíková involuce, hepatocelulární karcinom a zkrátil životnost.[16] Výzkum naznačuje, že zvýšené hladiny SOD1 by mohly být biomarkerem pro chronické toxicita těžkých kovů u žen s dlouhodobým účinkem zubní plomba výplně.[17]
Amyotrofická laterální skleróza (Lou Gehrigova choroba)
Mutace (více než 150 dosud identifikovaných) v tomto genu byly spojeny s familiárními Amyotrofní laterální skleróza.[18][19][20] Několik důkazů však také ukazuje, že divoký typ SOD1 je za podmínek buněčného stresu zapojen do významné části sporadických případů ALS, což představuje 90% pacientů s ALS.[21]Nejčastější mutace jsou A4V (v USA) a H46R (Japonsko). Pouze na Islandu SOD1-G93S bylo nalezeno. Nejvíce studovaný model myši ALS je G93A. U tohoto genu byly hlášeny vzácné varianty transkriptu.[10]
Prakticky všechny známé mutace SOD1 způsobující ALS působí v a dominantní móda; jediná mutantní kopie genu SOD1 je dostatečná k vyvolání onemocnění. Přesný molekulární mechanismus (nebo mechanismy), kterým mutace SOD1 způsobují onemocnění, není znám. Zdá se, že jde o nějaký toxický zisk funkce,[20] tolik mutantů SOD1 asociovaných s onemocněním (včetně G93A a A4V) si zachovává enzymatickou aktivitu a myši s knock-outem Sod1 nevyvíjejí ALS (i když vykazují silnou na věku závislou distální motorickou neuropatii).
ALS je neurodegenerativní onemocnění charakterizovaná selektivní ztrátou motorické neurony působit svalová atrofie. The Oxidace DNA produkt 8-OHdG je dobře zavedený ukazatel oxidační poškození DNA. 8-OHdG se hromadí v mitochondrie páteře motorické neurony osob s ALS.[22] v transgenní ALS myši nesoucí mutantní gen SOD1 se také hromadí 8-OHdG mitochondriální DNA spinálních motorických neuronů.[23] Tato zjištění naznačují, že oxidační poškození mitochondriální DNA motorických neuronů v důsledku změněného SOD1 může být významným faktorem v etiologii ALS.
Mutace A4V
A4V (alanin v kodonu 4 změněno na valin ) je nejčastější mutací způsobující ALS v americké populaci, přičemž přibližně 50% pacientů s SOD1-ALS nese mutaci A4V.[24][25][26] Přibližně 10 procent všech rodinných případů ALS v USA je způsobeno heterozygotními mutacemi A4V v SOD1. Mutace se zřídka, pokud vůbec, nachází mimo Ameriku.
Nedávno se odhadovalo, že mutace A4V nastala před 540 generacemi (~ 12 000 lety). Haplotyp obklopující mutaci naznačuje, že mutace A4V vznikla u asijských předků domorodých Američanů, kteří se dostali do Ameriky přes Beringova úžina.[27]
Mutant A4V patří k mutantům podobným WT. Pacienti s mutacemi A4V vykazují proměnlivý věk nástupu, ale rovnoměrně velmi rychlý průběh onemocnění, s průměrným přežitím po nástupu 1,4 roku (ve srovnání s 3–5 lety s jinými dominantními mutacemi SOD1, a v některých případech, jako je H46R, podstatně delší). Toto přežití je podstatně kratší než nemutantní ALS spojené s SOD1.
Mutace H46R
H46R (histidin na kodonu 47 změněno na arginin ) je nejčastější mutací způsobující ALS v japonské populaci, přičemž tuto mutaci nese přibližně 40% japonských pacientů s SOD1-ALS. H46R způsobuje hlubokou ztrátu vazby mědi v aktivním místě SOD1a jako takový je H46R enzymaticky neaktivní. Průběh této mutace je extrémně dlouhý, typický čas od nástupu do smrti je více než 15 let.[28] Myší modely s touto mutací nevykazují klasickou mitochondriální vakuolační patologii pozorovanou u myší G93A a G37R ALS a na rozdíl od myší G93A nedostatek hlavního mitochondriálního antioxidačního enzymu, SOD2, nemá žádný vliv na jejich průběh nemoci.[28]
Mutace G93A
G93A (glycin 93 změněn na alanin) je poměrně vzácná mutace, ale byla studována velmi intenzivně, protože to byla první mutace modelována u myší. G93A je pseudo-WT mutace, která ponechává aktivitu enzymu neporušená.[26] Z důvodu snadné dostupnosti myši G93A od Jacksonova laboratoř V tomto modelu bylo provedeno mnoho studií potenciálních cílů léčiv a mechanismů toxicity. Alespoň jeden soukromý výzkumný ústav (Institut rozvoje terapie ALS ) provádí rozsáhlé obrazovky s drogami výhradně v tomto modelu myši. Zda jsou nálezy specifické pro G93A nebo použitelné pro všechny ALS způsobující mutace SOD1, není v současnosti známo. Bylo argumentováno, že některé patologické rysy myši G93A jsou způsobeny artefakty nadměrné exprese, konkrétně těmi, které se týkají mitochondriální vakuolizace (myš G93A běžně používaná od Jackson Lab má přes 20 kopií lidského genu SOD1).[29] Alespoň jedna studie zjistila, že určité rysy patologie jsou idiosynkratické pro G93A a nelze je extrapolovat na všechny mutace způsobující ALS.[28] Další studie ukázaly, že patogeneze modelů G93A a H46R je jasně odlišná; některé léky a genetické intervence, které jsou v jednom modelu vysoce prospěšné / škodlivé, mají v druhém modelu opačný nebo žádný účinek.[30][31][32]
Downův syndrom
Downův syndrom (DS) je způsoben a triplikace chromozomu 21. Oxidační stres je považován za důležitý základní faktor v patologiích souvisejících s DS. Zdá se, že oxidační stres je způsoben triplikací a zvýšenou expresí genu SOD1 lokalizovaného v chromozomu 21. Zvýšená exprese SOD1 pravděpodobně způsobí zvýšenou produkci peroxid vodíku což vede ke zvýšenému poškození buněk.
Úrovně 8-OHdG v DNA osob s DS, měřeno v sliny Bylo zjištěno, že jsou významně vyšší než v kontrolních skupinách.[33] Hladiny 8-OHdG byly také zvýšeny v leukocyty osob s DS ve srovnání s kontrolami.[34] Tato zjištění naznačují, že poškození oxidační DNA může vést k některým klinickým rysům DS.
Interakce
SOD1 bylo prokázáno komunikovat s CCS[35] a Bcl-2.[36][37][38][39]
Reference
- ^ A b C GRCh38: Vydání souboru 89: ENSG00000142168 - Ensembl, Květen 2017
- ^ A b C GRCm38: Vydání souboru 89: ENSMUSG00000022982 - Ensembl, Květen 2017
- ^ „Human PubMed Reference:“. Národní centrum pro biotechnologické informace, Americká národní lékařská knihovna.
- ^ „Myš PubMed Reference:“. Národní centrum pro biotechnologické informace, Americká národní lékařská knihovna.
- ^ Milani P, Gagliardi S, Cova E, Cereda C (2011). „Transkripční a posttranskripční regulace SOD1 a její možné důsledky v ALS“. Neurology Research International. 2011: 1–9. doi:10.1155/2011/458427. PMC 3096450. PMID 21603028.
- ^ A b C Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX (březen 1993). „Mutace v genu superoxiddismutázy Cu / Zn jsou spojeny s familiární amyotrofickou laterální sklerózou“. Příroda. 362 (6415): 59–62. Bibcode:1993Natur.362 ... 59R. doi:10.1038 / 362059a0. PMID 8446170. S2CID 265436.
- ^ A b C Estácio SG, Leal SS, Cristóvão JS, Faísca PF, Gomes CM (únor 2015). „Vazba vápníku na zbytky vrátného, které lemují segmenty náchylné k agregaci, jsou základem nefibrilárních amyloidních znaků v superoxiddismutáze 1 (SOD1)“. Biochimica et Biophysica Acta (BBA) - bílkoviny a proteomika. 1854 (2): 118–26. doi:10.1016 / j.bbapap.2014.11.005. PMID 25463043.
- ^ A b C d Sea K, Sohn SH, Durazo A, Sheng Y, Shaw BF, Cao X, Taylor AB, Whitson LJ, Holloway SP, Hart PJ, Cabelli DE, Gralla EB, Valentine JS (leden 2015). „Pohledy na roli neobvyklé disulfidové vazby v superoxiddismutáze měď-zinek“. The Journal of Biological Chemistry. 290 (4): 2405–18. doi:10.1074 / jbc.M114.588798. PMC 4303690. PMID 25433341.
- ^ A b Khare SD, Caplow M, Dokholyan NV (říjen 2004). „Rychlostní a rovnovážné konstanty pro vícestupňovou reakční sekvenci agregace superoxiddismutázy při amyotrofické laterální skleróze“. Sborník Národní akademie věd Spojených států amerických. 101 (42): 15094–9. Bibcode:2004PNAS..10115094K. doi:10.1073 / pnas.0406650101. PMC 524068. PMID 15475574.
- ^ A b "Entrez Gene: SOD1 superoxid dismutáza 1, rozpustný (amyotrofická laterální skleróza 1 (dospělý))" ".
- ^ Murray CJ, Lopez AD (květen 1997). „Alternativní projekce úmrtnosti a zdravotního postižení podle příčin 1990–2020: Global Burden of Disease Study“. Lanceta. 349 (9064): 1498–504. doi:10.1016 / S0140-6736 (96) 07492-2. PMID 9167458. S2CID 10556268.
- ^ Braunwald E, Kloner RA (listopad 1985). „Reperfúze myokardu: dvojsečný meč?“. The Journal of Clinical Investigation. 76 (5): 1713–9. doi:10.1172 / JCI112160. PMC 424191. PMID 4056048.
- ^ Maslov LN, Naryzhnaia NV, Podoksenov IuK, Prokudina ES, Gorbunov AS, Zhang I, Peĭ ZhM (leden 2015). „[Reaktivní formy kyslíku jsou spouštěči a mediátory zvýšení srdeční tolerance k dopadu ischemie-reperfuze]“. Rossiĭskii Fiziologicheskiĭ Zhurnal Imeni I.M. Sechenova / Rossiĭskaia Akademiia Nauk. 101 (1): 3–24. PMID 25868322.
- ^ Liem DA, Honda HM, Zhang J, Woo D, Ping P (prosinec 2007). "Minulý a současný průběh kardioprotekce proti ischemicko-reperfuznímu poškození". Journal of Applied Physiology. 103 (6): 2129–36. doi:10.1152 / japplphysiol.00383.2007. PMID 17673563.
- ^ Udar N, Atilano SR, Brown DJ, Holguin B, Small K, Nesburn AB, Kenney MC (srpen 2006). „SOD1: kandidátský gen pro keratokonus“. Investigativní oftalmologie a vizuální věda. 47 (8): 3345–51. doi:10,1167 / iovs.05-1500. PMID 16877401.
- ^ Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H (srpen 2007). "Trendy v teoriích oxidačního stárnutí". Radikální biologie a medicína zdarma. 43 (4): 477–503. doi:10.1016 / j.freeradbiomed.2007.03.034. PMID 17640558.
- ^ Cabaña-Muñoz ME, Parmigiani-Izquierdo JM, Bravo-González LA, Kyung HM, Merino JJ (červen 2015). „Zvýšené hladiny zinku / glutathionu a vyšší aktivita superoxiddismutázy-1 jako biomarkery oxidačního stresu u žen s dlouhodobými zubními amalgámovými výplněmi: korelace mezi hladinami rtuti / hliníku (ve vlasech) a antioxidačními systémy v plazmě“. PLOS ONE. 10 (6): e0126339. Bibcode:2015PLoSO..1026339C. doi:10.1371 / journal.pone.0126339. PMC 4468144. PMID 26076368.
- ^ Conwit RA (prosinec 2006). „Prevence familiární ALS: klinické hodnocení může být proveditelné, ale je nutné provést hodnocení účinnosti?“. Časopis neurologických věd. 251 (1–2): 1–2. doi:10.1016 / j.jns.2006.07.009. PMID 17070848. S2CID 33105812.
- ^ Al-Chalabi A, Leigh PN (srpen 2000). "Nedávný pokrok v amyotrofické laterální skleróze". Aktuální názor v neurologii. 13 (4): 397–405. doi:10.1097/00019052-200008000-00006. PMID 10970056. S2CID 21577500.
- ^ A b Redler RL, Dokholyan NV (01.01.2012). "Komplexní molekulární biologie amyotrofické laterální sklerózy (ALS)". Molekulární biologie neurodegenerativních chorob. Pokrok v molekulární biologii a translační vědě. 107. 215–62. doi:10.1016 / B978-0-12-385883-2.00002-3. ISBN 9780123858832. PMC 3605887. PMID 22482452.
- ^ Gagliardi S, Cova E, Davin A, Guareschi S, Abel K, Alvisi E, Laforenza U, Ghidoni R, Cashman JR, Ceroni M, Cereda C (srpen 2010). "Exprese mRNA SOD1 u sporadické amyotrofické laterální sklerózy". Neurobiologie nemocí. 39 (2): 198–203. doi:10.1016 / j.nbd.2010.04.008. PMID 20399857. S2CID 207065284.
- ^ Kikuchi H, Furuta A, Nishioka K, Suzuki SO, Nakabeppu Y, Iwaki T (duben 2002). "Poškození mitochondriálních DNA opravných enzymů proti akumulaci 8-oxo-guaninu v spinálních motorických neuronech amyotrofické laterální sklerózy". Acta Neuropathol. 103 (4): 408–14. doi:10.1007 / s00401-001-0480-x. PMID 11904761. S2CID 2102463.
- ^ Warita H, Hayashi T, Murakami T, Manabe Y, Abe K (duben 2001). „Oxidační poškození mitochondriální DNA v spinálních motoneuronech transgenních ALS myší“. Brain Res. Mol. Brain Res. 89 (1–2): 147–52. doi:10.1016 / S0169-328X (01) 00029-8. PMID 11311985.
- ^ Rosen DR, Bowling AC, Patterson D, Usdin TB, Sapp P, Mezey E, McKenna-Yasek D, O'Regan J, Rahmani Z, Ferrante RJ (červen 1994). „Častá mutace ala 4 až val superoxid dismutázy-1 je spojena s rychle progresivní familiární amyotrofickou laterální sklerózou“. Lidská molekulární genetika. 3 (6): 981–7. doi:10,1093 / hmg / 3,6,981. PMID 7951249.
- ^ Cudkowicz ME, McKenna-Yasek D, Sapp PE, Chin W, Geller B, Hayden DL, Schoenfeld DA, Hosler BA, Horvitz HR, Brown RH (únor 1997). "Epidemiologie mutací v superoxiddismutáze při amyotrofické laterální skleróze". Annals of Neurology. 41 (2): 210–21. doi:10,1002 / analog. 410410212. PMID 9029070. S2CID 25595595.
- ^ A b Valentine JS, Hart PJ (duben 2003). „Špatně složený CuZnSOD a amyotrofická laterální skleróza“. Sborník Národní akademie věd Spojených států amerických. 100 (7): 3617–22. Bibcode:2003PNAS..100,3617V. doi:10.1073 / pnas.0730423100. PMC 152971. PMID 12655070.
- ^ Broom WJ, Johnson DV, Auwarter KE, Iafrate AJ, Russ C, Al-Chalabi A, Sapp PC, McKenna-Yasek D, Andersen PM, Brown RH (leden 2008). „ALS zprostředkovaná SOD1A4V: absence úzce spojeného modifikujícího genu a vznik v Asii“. Neurovědy Dopisy. 430 (3): 241–5. doi:10.1016 / j.neulet.2007.11.004. PMID 18055113. S2CID 46282375.
- ^ A b C Muller FL, Liu Y, Jernigan A, Borchelt D, Richardson A, Van Remmen H (září 2008). „Nedostatek MnSOD má rozdílný účinek na progresi onemocnění ve dvou různých myších modelech ALS mutantů“. Muscle & Nerve. 38 (3): 1173–83. doi:10,1002 / mus.21049. PMID 18720509. S2CID 23971601.
- ^ Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, Brännström T, Rehnmark A, Marklund SL (duben 2006). „Přetížení stabilních a vyloučení nestabilních variant lidské superoxiddismutázy-1 v mitochondriích myších modelů amyotrofické laterální sklerózy“. The Journal of Neuroscience. 26 (16): 4147–54. doi:10.1523 / JNEUROSCI.5461-05.2006. PMC 6673995. PMID 16624935.
- ^ Pan L, Yoshii Y, Otomo A, Ogawa H, Iwasaki Y, Shang HF, Hadano S (2012). „Různé mutanty humánní superoxidu měďno-zinečnatého dismutázy, SOD1G93A a SOD1H46R, mají zřetelné škodlivé účinky na celkový fenotyp u myší“. PLOS ONE. 7 (3): e33409. Bibcode:2012PLoSO ... 733409P. doi:10.1371 / journal.pone.0033409. PMC 3306410. PMID 22438926.
- ^ Bhattacharya A, Bokov A, Muller FL, Jernigan AL, Maslin K, Diaz V, Richardson A, Van Remmen H (srpen 2012). „Dietní omezení, ale ne rapamycin, prodlužuje nástup nemoci a přežití myšího modelu AL46 H46R / H48Q“. Neurobiologie stárnutí. 33 (8): 1829–32. doi:10.1016 / j.neurobiolaging.2011.06.002. PMID 21763036. S2CID 11227242.
- ^ Vargas MR, Johnson DA, Johnson JA (září 2011). „Snížený glutathion urychluje neurologický deficit a mitochondriální patologii u familiárního myšího modelu hSOD1 (G93A) spojeného s ALS“. Neurobiologie nemocí. 43 (3): 543–51. doi:10.1016 / j.nbd.2011.04.025. PMC 3139005. PMID 21600285.
- ^ Komatsu T, Duckyoung Y, Ito A, Kurosawa K, Maehata Y, Kubodera T, Ikeda M, Lee MC (září 2013). "Zvýšený biomarkery oxidačního stresu ve slinách pacientů s Downovým syndromem". Oblouk. Oral Biol. 58 (9): 1246–50. doi:10.1016 / j.archoralbio.2013.03.017. PMID 23714170.
- ^ Pallardó FV, Degan P, d'Ischia M, Kelly FJ, Zatterale A, Calzone R, Castello G, Fernandez-Delgado R, Dunster C, Lloret A, Manini P, Pisanti MA, Vuttariello E, Pagano G (srpen 2006). „Několik důkazů o prooxidačním stavu v raném věku u pacientů s Downovým syndromem“. Biogerontologie. 7 (4): 211–20. doi:10.1007 / s10522-006-9002-5. PMID 16612664. S2CID 13657691.
- ^ Casareno RL, Wagoner D, Gitlin JD (září 1998). „Chaperon mědi CCS přímo interaguje s superoxiddismutázou měď / zinek“. The Journal of Biological Chemistry. 273 (37): 23625–8. doi:10.1074 / jbc.273.37.23625. PMID 9726962.
- ^ Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH (červenec 2004). „S amyotrofickou laterální sklerózou asociované mutantní proteiny SOD1 se vážou a agregují s Bcl-2 v mitochondriích míchy“. Neuron. 43 (1): 19–30. doi:10.1016 / j.neuron.2004.06.021. PMID 15233914. S2CID 18141051.
- ^ Cova E, Ghiroldi A, Guareschi S, Mazzini G, Gagliardi S, Davin A, Bianchi M, Ceroni M, Cereda C (říjen 2010). „G93A SOD1 mění buněčný cyklus v buněčném modelu amyotrofické laterální sklerózy“. Mobilní signalizace. 22 (10): 1477–84. doi:10.1016 / j.cellsig.2010.05.016. PMID 20561900.
- ^ Cereda C, Cova E, Di Poto C, Galli A, Mazzini G, Corato M, Ceroni M (listopad 2006). „Vliv oxidu dusnatého na lymfocyty od pacientů se sporadickou amyotrofickou laterální sklerózou: toxická nebo ochranná role?“. Neurologické vědy. 27 (5): 312–6. doi:10.1007 / s10072-006-0702-z. PMID 17122939. S2CID 25059353.
- ^ Cova E, Cereda C, Galli A, Curti D, Finotti C, Di Poto C, Corato M, Mazzini G, Ceroni M (květen 2006). "Modifikovaná exprese proteinů Bcl-2 a SOD1 v lymfocytech od sporadických pacientů s ALS". Neurovědy Dopisy. 399 (3): 186–90. doi:10.1016 / j.neulet.2006.01.057. PMID 16495003. S2CID 26076370.
Další čtení
- de Belleroche J, Orrell R, král A (listopad 1995). „Familiární amyotrofická laterální skleróza / onemocnění motorických neuronů (FALS): přehled současného vývoje“. Journal of Medical Genetics. 32 (11): 841–7. doi:10,1136 / jmg.32.11.841. PMC 1051731. PMID 8592323.
- Ceroni M, Curti D, Alimonti D (2002). "Amyotrofická laterální skleróza a gen SOD1: přehled". Funkční neurologie. 16 (4 doplňky): 171–80. PMID 11996514.
- Zelko IN, Mariani TJ, Folz RJ (srpen 2002). „Multigenová rodina superoxiddismutázy: srovnání genových struktur, evoluce a exprese genů CuZn-SOD (SOD1), Mn-SOD (SOD2) a EC-SOD (SOD3)“. Radikální biologie a medicína zdarma. 33 (3): 337–49. doi:10.1016 / S0891-5849 (02) 00905-X. PMID 12126755.
- Hadano S (červen 2002). "[Příčinné geny pro familiární amyotrofickou laterální sklerózu]". Seikagaku. The Journal of Japanese Biochemical Society. 74 (6): 483–9. PMID 12138710.
- Noor R, Mittal S, Iqbal J (září 2002). "Superoxid dismutáza - aplikace a význam pro lidské nemoci". Monitor lékařské vědy. 8 (9): RA210–5. PMID 12218958.
- Potter SZ, Valentine JS (duben 2003). „Zmatená role superoxiddimutázy mědi a zinku v amyotrofické laterální skleróze (Lou Gehrigova choroba)“. Journal of Biological Anorganic Chemistry. 8 (4): 373–80. doi:10.1007 / s00775-003-0447-6. PMID 12644909. S2CID 22820101.
- Rotilio G, Aquilano K, Ciriolo MR (2004). "Souhra Cu, Zn superoxiddismutázy a syntázy oxidu dusnatého v neurodegenerativních procesech". IUBMB Life. 55 (10–11): 629–34. doi:10.1080/15216540310001628717. PMID 14711010. S2CID 19518719.
- Jafari-Schluep HF, Khoris J, Mayeux-Portas V, Hand C, Rouleau G, Camu W (leden 2004). „[Abnormality genů superoxyde dismutázy 1 u familiární amyotrofické laterální sklerózy: korelace fenotyp / genotyp. Francouzské zkušenosti a přehled literatury]“. Revue Neurologique. 160 (1): 44–50. doi:10.1016 / S0035-3787 (04) 70846-2. PMID 14978393.
- Faraci FM, Didion SP (srpen 2004). „Cévní ochrana: izoformy superoxiddismutázy ve stěně cévy“. Arterioskleróza, trombóza a vaskulární biologie. 24 (8): 1367–73. doi:10.1161 / 01.ATV.0000133604.20182.cf. PMID 15166009.
- Gagliardi S, Ogliari P, Davin A, Corato M, Cova E, Abel K, Cashman JR, Ceroni M, Cereda C (srpen 2011). „Hladiny mRNA monooxygenázy obsahující flavin jsou up-regulovány v oblastech mozku u myší s mutacemi SOD1“. Výzkum neurotoxicity. 20 (2): 150–8. doi:10.1007 / s12640-010-9230-r. PMID 21082301. S2CID 21856030.
- Battistini S, Ricci C, Lotti EM, Benigni M, Gagliardi S, Zucco R, Bondavalli M, Marcello N, Ceroni M, Cereda C (červen 2010). "Závažná familiární ALS s novou mutací exonu 4 (L106F) v genu SOD1". Časopis neurologických věd. 293 (1–2): 112–5. doi:10.1016 / j.jns.2010.03.009. PMID 20385392. S2CID 24895265.
Galerie PDB | |
|---|---|
|