Vícestavové modelování biomolekul - Multi-state modeling of biomolecules
Vícestavové modelování biomolekul odkazuje na řadu technik používaných k reprezentaci a výpočtu chování biologické molekuly nebo komplexy které mohou přijmout velké množství možných funkčních stavů.
Biologický signalizační systémy často spoléhají na biologické komplexy makromolekuly které mohou podstoupit několik funkčně významných modifikací, které jsou vzájemně kompatibilní. Mohou tedy existovat ve velmi velkém počtu funkčně odlišných stavů. Modelování takové vícestavové systémy představují dva problémy: Problém, jak popsat a specifikovat vícestavový systém („problém se specifikací“), a problém, jak pomocí počítače simulovat postup systému v čase („ výpočetní problém "). K řešení problému se specifikacemi se modeláři v posledních letech odklonili od explicitní specifikace všech možných stavů a směrem k na základě pravidel formalizmy, které umožňují implicitní specifikaci modelu, včetně κ-kalkulu,[1] BioNetGen,[2][3][4][5] Allosteric Network Compiler[6] a další.[7][8] Aby se vypořádali s problémem výpočtu, obrátili se na částicové metody, které se v mnoha případech ukázaly jako výpočetně účinnější než populační metody založené na obyčejné diferenciální rovnice, parciální diferenciální rovnice, nebo Gillespie stochastický simulační algoritmus.[9][10] Vzhledem k současné výpočetní technologii jsou metody založené na částicích někdy jedinou možnou možností. Simulátory založené na částicích dále spadají do dvou kategorií:prostorový simulátory jako StochSim,[11] DYNSTOC,[12] RuleMonkey,[9][13] a NFSim[14] a prostorové simulátory, včetně Meredys,[15] SRSim[16][17] a MCell.[18][19][20] Modeláři si tak mohou vybrat z různých nástrojů; nejlepší volba v závislosti na konkrétním problému. Probíhá vývoj rychlejších a výkonnějších metod, které slibují schopnost simulovat v budoucnu stále složitější signalizační procesy.
Úvod
Vícestupňové biomolekuly v signální transdukci
V životě buňky, signály jsou zpracovávány sítěmi bílkoviny které mohou fungovat jako složitá výpočetní zařízení.[21] Tyto sítě se spoléhají na schopnost jednotlivých proteinů existovat v různých funkčně odlišných stavech dosažených prostřednictvím různých mechanismů, včetně posttranslační úpravy, vazba ligandu, konformační změna, nebo tvorba nových komplexy.[21][22][23][24] Podobně, nukleové kyseliny může podstoupit řadu transformací, včetně vazby na proteiny, vazby jiných nukleových kyselin, konformačních změn a Methylace DNA.
Kromě toho může současně existovat několik typů modifikací, které mají kombinovaný vliv na biologickou makromolekulu v daném okamžiku. Biomolekula nebo komplex biomolekul může tedy často přijmout velmi velké množství funkčně odlišných stavů. Počet stavů se exponenciálně mění s počtem možných úprav, což je jev známý jako „kombinatorická exploze ".[24] To je znepokojující výpočetní biologové kdo takové biomolekuly modeluje nebo simuluje, protože vyvolává otázky o tom, jak lze reprezentovat a simulovat tak velký počet států.
Příklady kombinatorické exploze
Biologický signalizační sítě začlenit širokou škálu reverzibilních interakce, posttranslační úpravy a konformační změny. Navíc je běžné, že se protein skládá z několika - identických nebo neidentických - podjednotky a aby se několik proteinů a / nebo druhů nukleových kyselin shromáždilo do větších komplexů. Molekulární druh s několika z těchto znaků proto může existovat ve velkém počtu možných stavů.
Odhaduje se například, že droždí protein lešení Ste5 může být součástí 25666 jedinečných proteinových komplexů.[22] v E-coli, chemotaxe receptory čtyř různých druhů interagují ve skupinách po třech a každý jednotlivý receptor může existovat alespoň ve dvou možných konformacích a má až osm methylace weby,[23] což má za následek miliardy potenciálních stavů. Protein kináza CaMKII je dodecamer z dvanácti katalytické podjednotky,[25] uspořádány do dvou hexamerické prsteny.[26] Každá podjednotka může existovat alespoň ve dvou odlišných konformacích a každá podjednotka má různé vlastnosti fosforylace a vazebná místa pro ligand. Nedávný model[27] začleněné konformační stavy, dva fosforylace weby a dva způsoby vazby vápník / kalmodulin, tedy celkem kolem jedné miliardy možných stavů na hexamerický kruh. Model spojky EGF receptor do a MAP kináza kaskáda představená Danosem a kolegy[28] účty pro odlišné molekulární druhy, přesto si autoři všimli několika bodů, ve kterých by bylo možné model dále rozšířit. Novější model ErbB receptorová signalizace dokonce představuje více než jednu googol () odlišné molekulární druhy.[29] Problém kombinatorické exploze je také relevantní pro syntetická biologie, s nedávným modelem relativně jednoduchého syntetického materiálu eukaryotický genový obvod představovat 187 druhů a 1165 reakce.[30]


Samozřejmě, že ne všechny možné stavy vícestavové molekuly nebo komplexu budou nutně naplněny. Ve skutečnosti v systémech, kde je počet možných stavů mnohem větší než počet molekul v kompartmentu (např. Buňce), nemohou být. V některých případech lze empirické informace použít k vyloučení určitých stavů, pokud jsou například některé kombinace funkcí nekompatibilní. Při absenci takových informací je však třeba vzít v úvahu všechny možné státy a priori. V takových případech lze pomocí výpočetního modelování zjistit, do jaké míry jsou různé stavy naplněny.
Existence (nebo potenciální existence) tak velkého počtu molekulárních druhů je a kombinační fenomén: Vyplývá to z relativně malého souboru znaků nebo modifikací (jako je posttranslační modifikace nebo tvorba komplexu), které kombinují diktát stavu celé molekuly nebo komplexu, stejně jako existence jen několika možností v A kavárna (malé, střední nebo velké, s mlékem nebo bez mléka, káva bez kofeinu nebo ne, extra výstřel z espresso ) rychle vede k velkému počtu možných nápojů (v tomto případě 24; každá další binární volba toto číslo zdvojnásobí). I když je pro nás obtížné pochopit celkový počet možných kombinací, obvykle není koncepčně obtížné pochopit (mnohem menší) sadu znaků nebo modifikací a jejich vliv na funkci biomolekuly. Rychlost, jakou molekula podstoupí určitou reakci, bude obvykle záviset hlavně na jediném znaku nebo malé podskupině znaků. Je to přítomnost nebo nepřítomnost těch rysů, které diktují rychlost reakce. Reakční rychlost je stejná pro dvě molekuly, které se liší pouze vlastnostmi, které tuto reakci neovlivňují. Počet parametrů bude tedy mnohem menší než počet reakcí. (V příkladu kavárny bude přidání extra dávky espressa stát 40 centů, bez ohledu na velikost nápoje a bez ohledu na to, zda obsahuje mléko). Právě taková „místní pravidla“ se obvykle objevují při laboratorních experimentech. Multi-stavový model lze tedy konceptualizovat, pokud jde o kombinace modulárních funkcí a místních pravidel. To znamená, že ani model, který dokáže vysvětlit obrovské množství molekulárních druhů a reakcí, nemusí být nutně koncepčně složitý.
Specifikace vs. výpočet
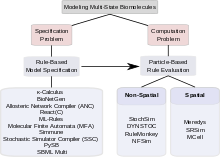
Kombinatorická složitost signalizačních systémů zahrnujících vícestavové proteiny představuje dva druhy problémů. První problém se týká toho, jak lze takový systém specifikovat; tj. jak může modelář robustním a efektivním způsobem specifikovat všechny komplexy, všechny změny, kterými tyto komplexy procházejí, a všechny parametry a podmínky, kterými se tyto změny řídí. Tento problém se nazývá „problém se specifikací“. Druhý problém se týká výpočet. Ptá se na to, zda je kombinatoricky složitý model, jakmile je specifikován, výpočetně využitelný, vzhledem k velkému počtu stavů a ještě většímu počtu možných přechodů mezi stavy, zda může být uložen elektronicky a zda jej lze vyhodnotit v rozumné míře množství výpočetního času. Tento problém se nazývá „výpočetní problém“. Mezi přístupy, které byly navrženy k řešení kombinatorické složitosti ve vícestavovém modelování, se některé zabývají hlavně řešením specifikačního problému, jiné se zaměřují na hledání účinných metod výpočtu. Některé nástroje se zabývají specifikacemi i výpočty. V následujících částech se diskutuje o pravidlových přístupech k problému se specifikací a o částicových přístupech k řešení problému výpočtu. Pro vícestavové modelování existuje široká škála výpočetních nástrojů.[31]
Problém se specifikací
Výslovná specifikace
Nejnaivnějším způsobem specifikování, např. Proteinu v biologickém modelu, je explicitně specifikovat každý z jeho stavů a použít každý z nich jako molekulární druh v simulace rámec, který umožňuje přechody ze státu do státu. Například pokud protein může být ligand -bound nebo ne, existují ve dvou konformační stavy (např. otevřené nebo uzavřené) a být umístěny ve dvou možných subcelulárních oblastech (např. cytosolický nebo membrána -bound), pak lze osm možných výsledných stavů explicitně vyjmenovat jako:
- vázaný, otevřený, cytosol
- vázaná, otevřená, membrána
- vázaný, uzavřený, cytosol
- vázané, uzavřené, membrány
- nevázané, otevřené, cytosol
- nevázaná, otevřená, membrána
- nevázané, uzavřené, cytosol
- nevázané, uzavřené, membrána
Výčet všech možných stavů je zdlouhavý a potenciálně náchylný k chybám. U makromolekulárních komplexů, které mohou přijmout více stavů, je výčet každého stavu rychle zdlouhavý, ne-li nemožný. Navíc přidání jedné další modifikace nebo funkce k modelu komplexu, který je předmětem vyšetřování, zdvojnásobí počet možných stavů (pokud je modifikace binární) a více než zdvojnásobí počet přechodů, které je třeba specifikovat.
Specifikace modelu na základě pravidel
Je zřejmé, že explicitní popis, který uvádí všechny možné molekulární druhy (včetně všech jejich možných stavů), všechny možné reakce nebo přechody, které mohou tyto druhy podstoupit, a všechny parametry, jimiž se tyto reakce řídí, se velmi rychle stává nepraktickým, protože složitost biologického systému zvyšuje. Modeláři proto hledali implicitní, spíše než explicitní způsoby specifikace biologického signalizačního systému. Implicitní popis je ten, který seskupuje reakce a parametry, které platí pro mnoho typů molekulárních druhů do jedné reakční šablony. Může také přidat soubor podmínek, které určují parametry reakce, tj. Pravděpodobnost nebo rychlost reakce, nebo zda k ní vůbec dojde. Pouze vlastnosti molekuly nebo komplexu, které jsou pro danou reakci důležité (ať už ovlivňují reakci nebo jsou jimi ovlivňovány), jsou výslovně uvedeny a všechny ostatní vlastnosti jsou ve specifikaci reakce ignorovány.
Například rychlost ligandu disociace z proteinu může záviset na konformačním stavu proteinu, ale ne na jeho subcelulární lokalizaci. Implicitní popis by proto uváděl dva disociační procesy (s různými rychlostmi v závislosti na konformačním stavu), ale ignoroval by atributy odkazující na subcelulární lokalizaci, protože neovlivňují rychlost disociace ligandu, ani nejsou ovlivněny. Toto pravidlo specifikace bylo shrnuto jako „Nezajímejte se, nepište“.[28]
Protože to není psáno ve smyslu reakcí, ale ve smyslu obecnějších „reakčních pravidel“ zahrnujících sady reakcí, tento druh specifikace se často nazývá „na základě pravidel“.[4] Tento popis systému, pokud jde o modulární pravidla, se opírá o předpoklad, že pro konkrétní pravidlo reakce je relevantní pouze podmnožina funkcí nebo atributů. Pokud platí tento předpoklad, může být sada reakcí hrubozrnná do jednoho reakčního pravidla. Toto hrubé zrnění zachovává důležité vlastnosti základních reakcí. Například pokud jsou reakce založeny na chemické kinetice, jsou odvozena i pravidla z nich odvozená.
Existuje mnoho metod specifikace založených na pravidlech. Obecně je specifikace modelu samostatným úkolem od provedení simulace. Proto mezi stávajícími systémy specifikací modelů založených na pravidlech[4] některé se soustředí pouze na specifikaci modelu, což uživateli umožňuje exportovat zadaný model do vyhrazeného simulačního nástroje. Mnoho řešení problému se specifikací však také obsahuje metodu interpretace zadaného modelu.[3] To se provádí poskytnutím metody simulace modelu nebo metody převodu do formy, kterou lze použít pro simulace v jiných programech.
Časná metoda specifikace založená na pravidlech je κ-kalkul,[1] A zpracovat algebru které lze použít ke kódování makromolekul s vnitřními stavy a vazebnými místy a k určení pravidel, podle kterých interagují.[28] Κ-kalkul se zabývá pouze poskytováním jazyka pro kódování vícestavových modelů, nikoli interpretací samotných modelů. Simulátor kompatibilní s Kappa je KaSim.[32][33]
BioNetGen je softwarová sada, která poskytuje kapacitu pro specifikaci i simulaci.[2][3][4][5] Modely založené na pravidlech lze zapisovat pomocí zadané syntaxe, jazyka BioNetGen (BNGL).[4] Základní koncepcí je reprezentovat biochemické systémy jako grafy, kde jsou molekuly reprezentovány jako uzly (nebo kolekce uzlů) a chemické vazby jako hrany. Pravidlo reakce pak odpovídá pravidlu přepisování grafu.[3] BNGL poskytuje syntaxi pro specifikaci těchto grafů a souvisejících pravidel jako strukturovaných řetězců.[4] BioNetGen pak může pomocí těchto pravidel generovat běžné diferenciální rovnice (ODE) k popisu každé biochemické reakce. Alternativně může generovat seznam všech možných druhů a reakcí v SBML,[34][35] které lze poté exportovat do simulačních softwarových balíčků, které umí číst SBML. Lze také využít vlastní simulační software založený na ODE společnosti BioNetGen a jeho schopnost generovat reakce během letu během stochastické simulace.[5] Model specifikovaný v BNGL lze navíc číst jiným simulačním softwarem, jako je DYNSTOC,[12] RuleMonkey,[13] a NFSim.[14]
Dalším nástrojem, který generuje plné reakční sítě ze sady pravidel, je Allosteric Network Compiler (ANC).[6] Koncepčně ANC vidí molekuly jako alosterická zařízení s a Monod-Wyman-Changeux (MWC) regulační mechanismus typu,[36] jejichž interakce se řídí jejich vnitřním stavem i vnějšími úpravami. Velmi užitečnou funkcí ANC je, že automaticky počítá závislé parametry, čímž ukládá termodynamické správnost.[37]
Rozšíření k-kalkulu poskytuje Reagovat (C).[38] Autoři Reagovat C. ukázat, že dokáže vyjádřit stochastický počet π.[39] Poskytují také stochastický simulační algoritmus založený na Gillespieho stochastickém algoritmu [40] pro modely uvedené v Reagovat (C).[38]
Pravidla ML[41] je podobný React (C), ale poskytuje přidanou možnost vnoření: Druh komponent modelu, se všemi jeho atributy, může být součástí druhů komponent vyššího řádu. To umožňuje ML-Rules zachytit víceúrovňové modely, které mohou překlenout propast mezi například řadou biochemických procesů a makroskopickým chováním celé buňky nebo skupiny buněk. Například model konceptu buněčného dělení v proof-of-concept štěpné droždí zahrnuje cyklin /cdc2 vazba a aktivace, feromon sekrece a difúze, buněčné dělení a pohyb buněk.[41] Modely uvedené v pravidlech ML lze simulovat pomocí simulačního rámce Jamese II.[42] Podobný vnořený jazyk, který představuje víceúrovňové biologické systémy, navrhli Oury a Plotkin.[43] Formalizmus specifikace založený na molekulárním konečné automaty Rámec (MFA) lze poté použít ke generování a simulaci systému ODR nebo pro stochastická simulace pomocí kinetiky Monte Carlo algoritmus.[8]
Některé systémy specifikací založené na pravidlech a jejich přidružené nástroje pro generování a simulaci sítí byly navrženy tak, aby vyhovovaly prostorové heterogenitě, aby bylo možné realistickou simulaci interakcí v biologických kompartmentech. Například projekt Simmune[44][45] zahrnuje prostorovou složku: Uživatelé mohou specifikovat své vícestavové biomolekuly a interakce v membránách nebo oddílech libovolného tvaru. Reakční objem je poté rozdělen na vzájemně propojené voxely a pro každý z těchto dílčích objemů je vytvořena samostatná reakční síť.
Stochastic Simulator Compiler (SSC)[46] umožňuje na základě pravidel modulární specifikaci interagujících biomolekul v oblastech libovolně složitých geometrií. Systém je opět znázorněn pomocí grafů, přičemž chemické interakce nebo difúzní události jsou formalizovány jako pravidla přepisování grafů.[46] Kompilátor poté vygeneruje celou reakční síť před spuštěním stochastického algoritmu reakce a difúze.
PySB používá jiný přístup,[47] kde je specifikace modelu zabudována do programovacího jazyka Krajta. Model (nebo jeho část) je reprezentován jako program v Pythonu. To umožňuje uživatelům ukládat biochemické procesy vyššího řádu, jako je katalýza nebo polymerace jako makra a podle potřeby je znovu použijte. Modely lze simulovat a analyzovat pomocí knihoven Pythonu, ale modely PySB lze také exportovat do BNGL,[4] kappa,[1] a SBML.[34]
Modely zahrnující vícestavové a vícesložkové druhy lze také specifikovat na úrovni 3 v System Biology Markup Language (SBML) [34] pomocí vícenásobného balíčku. K dispozici je návrh specifikace,[48] a softwarová podpora je ve vývoji.
Tím, že vezmeme v úvahu pouze stavy a vlastnosti důležité pro konkrétní reakci, specifikace modelu založená na pravidlech eliminuje potřebu výslovně vyjmenovat každý možný molekulární stav, který může podstoupit podobnou reakci, a tím umožňuje efektivní specifikaci.
Problém výpočtu
Při běhu simulace na biologickém modelu jakýkoli simulační software vyhodnotí soubor pravidel, počínaje od zadaného souboru počátečních podmínek, a obvykle iterace prostřednictvím řady časových kroků až do zadaného času ukončení. Jedním ze způsobů, jak klasifikovat simulační algoritmy, je podívat se na úroveň analýzy, na které se pravidla uplatňují: mohou být založené na populaci, založené na jedné částice nebo hybridní.
Populační hodnocení pravidel
Při hodnocení pravidel založených na populaci se pravidla aplikují na populace. Všechno molekuly stejné druh ve stejném stavu jsou spojeny dohromady. Použití konkrétního pravidla zmenšuje nebo zvětšuje velikost jednoho z fondů, možná na úkor jiného.
Některé z nejznámějších tříd simulačních přístupů ve výpočetní biologii patří do populační rodiny, včetně těch založených na numerické integraci běžných a parciálních diferenciálních rovnic a Gillespieho stochastického simulačního algoritmu.
Diferenciální rovnice popsat změny molekulárních koncentrací v čase deterministickým způsobem. Simulace založené na diferenciálních rovnicích se obvykle nepokouší řešit tyto rovnice analyticky, ale používají vhodné numerický řešič.
Stochastický Gillespieho algoritmus mění složení směsí molekul v průběhu náhodnost reakční události, pravděpodobnost z nichž se počítá z reakčních rychlostí a z počtu molekul v souladu se stochastickou hlavní rovnice.[40]
V populačních přístupech lze uvažovat o modelovaném systému jako o daném stavu v kterémkoli daném časovém bodě, kde je stav definován podle povahy a velikosti osídlených skupin molekul. To znamená, že prostor všech možných stavů může být velmi velký. U některých simulačních metod implementujících numerickou integraci běžných a parciálních diferenciálních rovnic nebo Gillespieho stochastického algoritmu jsou na začátku simulace definovány všechny možné zásoby molekul a reakce, kterými procházejí, i když jsou prázdné. Takové metody „generovat první“[4] měřítko špatně s rostoucím počtem molekulárních stavů.[49] Například se nedávno odhadovalo, že i pro jednoduchý model CaMKII s pouhými 6 stavy na podjednotky a 10 podjednotkami by trvalo generování celé reakční sítě na 2,54 GHz procesoru Intel 290 let Xeon procesor.[50] Kromě toho krok generování modelu v metodách generování první nemusí nutně končit, například když model zahrnuje sestavení proteinů do komplexů libovolně velké velikosti, jako je například aktin vlákna. V těchto případech musí být podmínka ukončení specifikována uživatelem.[3][5]
I když lze úspěšně vygenerovat velký reakční systém, jeho simulace s využitím vyhodnocení pravidel založených na populaci může narazit na výpočetní limity. V nedávné studii bylo prokázáno, že výkonný počítač není schopen simulovat protein s více než 8 fosforylace weby ( stavy fosforylace) pomocí obyčejných diferenciálních rovnic.[14]

Byly navrženy metody ke zmenšení velikosti stavového prostoru. Jedním z nich je zvážit v každém časovém bodě pouze stavy sousedící s aktuálním stavem (tj. Stavy, kterých lze dosáhnout v rámci další iterace). To eliminuje potřebu výčtu všech možných stavů na začátku. Místo toho jsou reakce generovány „za běhu“[4] při každé iteraci. Tyto metody jsou k dispozici jak pro stochastické, tak pro deterministické algoritmy. Tyto metody stále spoléhají na definici (i když omezené) reakční sítě - na rozdíl od níže popsaných „bezsíťových“ metod.
I při generování „on-the-fly“ sítí se sítě generované pro vyhodnocení pravidel založených na populaci mohou stát poměrně velkými, a proto je obtížné - ne-li nemožné - výpočetně je zpracovat. Alternativní přístup poskytuje hodnocení pravidel na základě částic.
Hodnocení pravidel na základě částic

V částicových (někdy nazývaných „agent-based“) simulacích proteiny, nukleové kyseliny, makromolekulární komplexy nebo malé molekuly jsou reprezentovány jako individuální software předměty a jejich pokrok je sledován v průběhu celé simulace.[51] Protože hodnocení pravidel založených na částicích sleduje spíše jednotlivé částice než populace, přichází při modelování systémů s vysokým celkovým počtem částic, ale malým počtem druhů (nebo skupin) částic, vyšší výpočetní náklady.[51] V případech kombinatorické složitosti je však modelování jednotlivých částic výhodou, protože v kterémkoli daném bodě simulace je třeba brát v úvahu pouze existující molekuly, jejich stavy a reakce, které mohou podstoupit. Hodnocení pravidel založených na částicích nevyžaduje generování úplných nebo částečných reakčních sítí na začátku simulace nebo v jakémkoli jiném bodě simulace, a proto se nazývá „bez sítě“.
Tato metoda snižuje složitost modelu ve fázi simulace, a tím šetří čas a výpočetní výkon.[9][10] Simulace sleduje každou částici a v každém simulačním kroku částice pouze „vidí“ reakce (nebo pravidla), které se na ni vztahují. To závisí na stavu částice a v některých implementacích na stavech jejích sousedů v holoenzymu nebo komplexu. Jak simulace pokračuje, stavy částic se aktualizují podle pravidel, která se aktivují.[10]
Některé částicové simulační balíčky používají ad hoc formalismus pro specifikaci reaktantů, parametrů a pravidel. Ostatní mohou číst soubory v uznávaném formátu specifikace založeném na pravidlech, jako je BNGL.[4]
Neprostorové metody založené na částicích
StochSim[11][52] je na bázi částic stochastický simulátor používaný hlavně k modelování chemických reakcí a jiných molekulárních přechodů. Algoritmus použitý v programu StochSim se liší od známějšího stochastického algoritmu Gillespie[40] v tom, že funguje na jednotlivých entitách, nikoli na seskupeních entit, takže je spíše na bázi částic než na populaci.
Ve StochSim může být každý molekulární druh vybaven řadou binárních stavů vlajky představující konkrétní modifikaci. Reakce mohou být závislé na sadě stavových příznaků nastavených na konkrétní hodnoty. Výsledek reakce může navíc zahrnovat změnu příznaku stavu. Subjekty lze navíc uspořádat geometricky pole (například pro holoenzymy skládající se z několika podjednotek) a reakce mohou být „citlivé na souseda“, tj. pravděpodobnost reakce pro danou entitu je ovlivněna hodnotou státní vlajky na sousední entitě. Díky těmto vlastnostem je StochSim ideální pro modelování vícestavových molekul uspořádaných do holoenzymů nebo komplexů specifikované velikosti. StochSim byl skutečně použit k modelování shluků bakteriální chemotaktický receptory,[53] a CaMKII holoenzymy.[27]
Rozšíření StochSim zahrnuje částicový simulátor DYNSTOC, který používá algoritmus podobný StochSim k simulaci modelů specifikovaných v jazyce BioNetGen (BNGL),[4] a zlepšuje zacházení s molekulami uvnitř makromolekulární komplexy.[12]
Dalším stochastickým simulátorem založeným na částicích, který umí číst vstupní soubory BNGL, je RuleMonkey.[13] Jeho simulační algoritmus[9] se liší od algoritmů, na nichž stojí StochSim i DYNSTOC, tím, že časový krok simulace je variabilní.
Network-Free Stochastic Simulator (NFSim) se liší od výše popsaných tím, že umožňuje definovat reakční rychlosti jako libovolné matematické nebo podmíněné výrazy, a tím usnadňuje selektivní hrubozrnný modelů.[14] RuleMonkey a NFsim implementují odlišné, ale související simulační algoritmy. Podrobný přehled a srovnání obou nástrojů uvádějí Yang a Hlavacek.[54]
Je snadné si představit biologický systém, kde některé komponenty jsou složité vícestavové molekuly, zatímco jiné mají několik možných stavů (nebo dokonce jen jeden) a existují ve velkém počtu. Pro modelování těchto systémů byl navržen hybridní přístup: V rámci hybridní částice / populace (HPP) může uživatel určit model založený na pravidlech, ale může určit některé druhy, které mají být v následných případech považovány za populace (spíše než částice). simulace.[10] Tato metoda kombinuje výpočetní výhody modelování založeného na částicích pro vícestavové systémy s relativně nízkým počtem molekul a populačního modelování pro systémy s vysokým počtem molekul a malým počtem možných stavů. Specifikace modelů HPP je podporována programem BioNetGen,[4] a simulace lze provádět pomocí NFSim.[14]
Metody založené na prostorových částicích

Metody založené na prostorových částicích se liší od metod popsaných výše jejich explicitním znázorněním prostoru.
Jedním příkladem simulátoru založeného na částicích, který umožňuje reprezentaci celulárních oddílů, je SRSim.[16][17] SRSim je integrován do simulátoru molekulární dynamiky LAMMPS[56][57] a umožňuje uživateli určit model v BNGL.[4] SRSim umožňuje uživatelům specifikovat geometrii částic v simulaci i stránky interakce. Proto je obzvláště dobré simulovat sestavu a strukturu komplexních biomolekulárních komplexů, o čemž svědčí nedávný model vnitřního kinetochore.[58]
MCell[18][19][20][59] umožňuje sledování jednotlivých molekul v libovolně složitých geometrických prostředích, která jsou definována uživatelem. To umožňuje simulace biomolekul v realistické rekonstrukci živých buněk, včetně buněk se složitou geometrií, jako jsou ty z neurony. Reakční komora je rekonstrukcí dendritické páteře.[55] Vizualizace jsou podporovány specializovaným modulem plug-in („CellBlender“) pro open source program Blender.[60]
MCell používá ad hoc formalismus v samotném MCell k určení multi-stavového modelu: V MCell je možné přiřadit „sloty“ libovolnému molekulární druhy. Každý slot znamená konkrétní modifikaci a molekule lze přiřadit libovolný počet slotů. Každý slot může být obsazen konkrétním státem. Státy nemusí být nutně binární. Například slot popisující vazbu konkrétního ligand k proteinu, který nás zajímá, může nabrat stavy „nevázaný“, „částečně vázaný“ a „plně vázaný“.
Syntaxi slotu a stavu v MCell lze také použít k modelování multimerních proteinů nebo makromolekulárních komplexů. Při použití tímto způsobem je slot zástupný symbol pro podjednotku nebo molekulární složku a komplex a stav štěrbiny bude indikovat, zda konkrétní proteinová složka chybí nebo je přítomna v komplexu. Způsob, jak o tom přemýšlet, je, že makromolekuly MCell mohou mít několik rozměry: „Stavová dimenze“ a jedna nebo více „prostorových dimenzí“. „Stavová dimenze“ se používá k popisu více možných stavů tvořících vícestavový protein, zatímco prostorová dimenze popisuje topologické vztahy mezi sousedními podjednotkami nebo členy makromolekulárního komplexu. Jednou z nevýhod této metody reprezentace proteinových komplexů ve srovnání s Meredysem je to, že MCell neumožňuje difúze komplexů, a tedy i vícestavových molekul. To lze v některých případech obejít úpravou difuzních konstant ligandů, které interagují s komplexem, pomocí funkcí kontrolního bodu nebo kombinací simulací na různých úrovních.
Příklady vícestavových modelů v biologii
(V žádném případě není vyčerpávající) výběr modelů biologických systémů zahrnujících vícestavové molekuly a za použití některých zde diskutovaných nástrojů je uveden v tabulce níže.
| Biologický systém | Specifikace | Výpočet | Odkaz | |
|---|---|---|---|---|
| Signální dráha bakteriální chemotaxe | StochSim | StochSim | [61] | |
| Regulace CaMKII | StochSim | StochSim | [27] | |
| ERBB signalizace receptoru | BioNetGen | NFSim | [29] | |
| Obvody eukaryotických syntetických genů | BioNetGen, PROPAGACE[62] | COPASI[63] | [30] | |
| RNA signalizace | Kappa | KaSim | [64] | |
| Spolupráce alosterických proteinů | Allosteric Network Compiler (ANC) | MATLAB | [6] | |
| Chemosenzování v Dictyostelium | Simmune | Simmune | [44] | |
| Receptor T-buněk aktivace | SSC | SSC | [65] | |
| Lidský mitotický kinetochor | BioNetGen | SRSim | [66] | |
| Buněčný cyklus štěpných kvasinek | Pravidla ML | JAMES II[42] | [41] |
Viz také
Reference
![]() Tento článek byl upraven z následujícího zdroje pod a CC BY 4.0 licence (2014 ) (zprávy recenzenta ): "Multi-state modeling of biomolecules", PLOS výpočetní biologie, 10 (9): e1003844, září 2014, doi:10.1371 / JOURNAL.PCBI.1003844, ISSN 1553-734X, PMC 4201162, PMID 25254957, Wikidata Q18145441
Tento článek byl upraven z následujícího zdroje pod a CC BY 4.0 licence (2014 ) (zprávy recenzenta ): "Multi-state modeling of biomolecules", PLOS výpočetní biologie, 10 (9): e1003844, září 2014, doi:10.1371 / JOURNAL.PCBI.1003844, ISSN 1553-734X, PMC 4201162, PMID 25254957, Wikidata Q18145441
- ^ A b C Danos, V; Laneve, C (2004). "Formální molekulární biologie". Teoretická informatika. 325: 69–110. doi:10.1016 / j.tcs.2004.03.065.
- ^ A b Blinov, M.L .; Faeder, J. R .; Goldstein, B; Hlaváček, W. S. (2004). "Bio Síť Gen: Software pro modelování signální transdukce založené na pravidlech na základě interakcí molekulárních domén ". Bioinformatika. 20 (17): 3289–91. doi:10.1093 / bioinformatika / bth378. PMID 15217809.
- ^ A b C d E Faeder, JR; Blinov, ML; Goldstein, B; Hlaváček, ZS (2005). „Modelování biochemických sítí na základě pravidel“. Složitost. 10 (4): 22–41. Bibcode:2005Cmplx..10d..22F. doi:10.1002 / cplx.20074. S2CID 9307441.
- ^ A b C d E F G h i j k l m Hlaváček, W. S .; Faeder, J. R .; Blinov, M.L .; Posner, R. G .; Hucka, M; Fontana, W (2006). "Pravidla pro modelování systémů přenosu signálu". Vědecká signalizace. 2006 (344): re6. CiteSeerX 10.1.1.83.1561. doi:10.1126 / stke.3442006re6. PMID 16849649. S2CID 1816082.
- ^ A b C d Faeder, J. R .; Blinov, M.L .; Hlaváček, W. S. (2009). Pravidlové modelování biochemických systémů s Bio SíťGen. Metody v molekulární biologii. 500. 113–67. CiteSeerX 10.1.1.323.9577. doi:10.1007/978-1-59745-525-1_5. ISBN 978-1-934115-64-0. PMID 19399430.
- ^ A b C Ollivier, J. F .; Shahrezaei, V; Swain, P. S. (2010). „Škálovatelné modelování alosterických proteinů a biochemických sítí na základě pravidel“. PLOS výpočetní biologie. 6 (11): e1000975. Bibcode:2010PLSCB ... 6E0975O. doi:10,1371 / journal.pcbi.1000975. PMC 2973810. PMID 21079669.
- ^ Lok, L; Brent, R (2005). "Automatické generování celulárních reakčních sítí s Moleculizer 1.0". Přírodní biotechnologie. 23 (1): 131–6. doi:10.1038 / nbt1054. PMID 15637632. S2CID 23696958.
- ^ A b Yang, J; Meng, X; Hlaváček, W. S. (2010). „Pravidlové modelování a simulace biochemických systémů s molekulárně konečnými automaty“. Biologie systémů IET. 4 (6): 453–66. arXiv:1007.1315. doi:10.1049 / iet-syb.2010.0015. PMC 3070173. PMID 21073243.
- ^ A b C d Yang, J; Monine, M. I .; Faeder, J. R .; Hlaváček, W. S. (2008). „Metoda kinetického Monte Carla pro modelování biochemických sítí na základě pravidel“. Fyzický přehled E. 78 (3 Pt 1): 031910. arXiv:0712.3773. Bibcode:2008PhRvE..78c1910Y. doi:10.1103/PhysRevE.78.031910. PMC 2652652. PMID 18851068.
- ^ A b C d Hogg, J. S., Harris, L. A., Stover, L. J., Nair, N. S., & Faeder, J. R. (2013). Exact hybrid particle/population simulation of rule-based models of biochemical systems. arXiv preprint arXiv:1301.6854.
- ^ A b Nov, Le; Shimizu, TS (2001). "STOCHSIM: modelling of stochastic biomolecular processes". Bioinformatika. 17 (6): 575–576. doi:10.1093/bioinformatics/17.6.575. PMID 11395441.
- ^ A b C Colvin, J; Monine, M. I.; Faeder, J. R.; Hlavacek, W. S.; von Hoff, D. D.; Posner, R. G. (2009). "Simulation of large-scale rule-based models". Bioinformatika. 25 (7): 910–7. doi:10.1093/bioinformatics/btp066. PMC 2660871. PMID 19213740.
- ^ A b C Colvin, J; Monine, M. I.; Gutenkunst, R. N.; Hlavacek, W. S.; von Hoff, D. D.; Posner, R. G. (2010). "Rule Opice: Software for stochastic simulation of rule-based models". BMC bioinformatika. 11: 404. doi:10.1186/1471-2105-11-404. PMC 2921409. PMID 20673321.
- ^ A b C d E Sneddon, M. W.; Faeder, J. R.; Emonet, T (2011). "Efficient modeling, simulation and coarse-graining of biological complexity with NFsim". Přírodní metody. 8 (2): 177–83. doi:10.1038/nmeth.1546. PMID 21186362. S2CID 5412795.
- ^ Tolle, D. P.; Le Novère, N (2010). "Meredys, a multi-compartment reaction-diffusion simulator using multistate realistic molecular complexes". Biologie systémů BMC. 4: 24. doi:10.1186/1752-0509-4-24. PMC 2848630. PMID 20233406.
- ^ A b Gruenert, G; Ibrahim, B; Lenser, T; Lohel, M; Hinze, T; Dittrich, P (2010). "Rule-based spatial modeling with diffusing, geometrically constrained molecules". BMC bioinformatika. 11: 307. doi:10.1186/1471-2105-11-307. PMC 2911456. PMID 20529264.
- ^ A b Grunert G, Dittrich P (2011) Using the SRSim Software for Spatial and Rule-Based Modeling of Combinatorially Complex Biochemical Reaction Systems. Membrane Computing - Lecture Notes in Computer Science 6501:240-256
- ^ A b Stiles, J. R.; Van Helden, D; Bartol Jr, T. M.; Salpeter, E. E.; Salpeter, M. M. (1996). "Miniature endplate current rise times less than 100 microseconds from improved dual recordings can be modeled with passive acetylcholine diffusion from a synaptic vesicle". Sborník Národní akademie věd Spojených států amerických. 93 (12): 5747–52. Bibcode:1996PNAS...93.5747S. doi:10.1073/pnas.93.12.5747. PMC 39132. PMID 8650164.
- ^ A b Stiles JR, Bartol TM (2001). Computational Neuroscience: Realistic Modeling for Experimentalists. In: De Schutter, E (ed). Computational Neuroscience: Realistic Modeling for Experimentalists. CRC Press, Boca Raton.
- ^ A b Kerr, R. A.; Bartol, T. M.; Kaminsky, B; Dittrich, M; Chang, J. C .; Baden, S. B.; Sejnowski, T. J.; Stiles, J. R. (2008). "Fast Monte Carlo Simulation Methods for Biological Reaction-Diffusion Systems in Solution and on Surfaces". SIAM Journal on Scientific Computing. 30 (6): 3126–3149. doi:10.1137/070692017. PMC 2819163. PMID 20151023.
- ^ A b Bray, D (1995). "Protein molecules as computational elements in living cells". Příroda. 376 (6538): 307–12. Bibcode:1995Natur.376..307B. doi:10.1038/376307a0. PMID 7630396. S2CID 4326068.
- ^ A b Endy, D.; Brent, R. (2001). "Modelling cellular behaviour". Příroda. 409 (6818): 391–395. Bibcode:2001Natur.409..391E. doi:10.1038/35053181. PMID 11201753. S2CID 480515.
- ^ A b Bray, D (2003). "Genomics. Molecular prodigality". Věda. 299 (5610): 1189–90. doi:10.1126/science.1080010. PMID 12595679. S2CID 34035288.
- ^ A b Hlavacek, W. S.; Faeder, J. R.; Blinov, M. L.; Perelson, A. S.; Goldstein, B (2003). "The complexity of complexes in signal transduction". Biotechnologie a bioinženýrství. 84 (7): 783–94. doi:10.1002/bit.10842. PMID 14708119. S2CID 9092264.
- ^ Bennett, M. K.; Erondu, N. E.; Kennedy, M. B. (1983). "Purification and characterization of a calmodulin-dependent protein kinase that is highly concentrated in brain". The Journal of Biological Chemistry. 258 (20): 12735–44. PMID 6313675.
- ^ Rosenberg, O. S.; Deindl, S; Sung, R. J .; Nairn, A. C.; Kuriyan, J (2005). "Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme". Buňka. 123 (5): 849–60. doi:10.1016/j.cell.2005.10.029. PMID 16325579. S2CID 2654357.
- ^ A b C Stefan, M. I.; Marshall, D. P.; Le Novère, N (2012). "Structural analysis and stochastic modelling suggest a mechanism for calmodulin trapping by CaMKII". PLOS ONE. 7 (1): e29406. Bibcode:2012PLoSO...729406S. doi:10.1371/journal.pone.0029406. PMC 3261145. PMID 22279535.
- ^ A b C Danos V, Feret J, Fontana W, Harmer R, Krivine J (2007). Rule-Based Modelling of Cellular Signalling. Proceedings of the Eighteenth International Conference on Concurrency Theory, CONCUR 2007, Lisbon, Portugal
- ^ A b Creamer, M. S.; Stites, E. C.; Aziz, M; Cahill, J. A.; Tan, C. W.; Berens, M. E.; Han, H; Bussey, K. J.; von Hoff, D. D.; Hlavacek, W. S.; Posner, R. G. (2012). "Specification, annotation, visualization and simulation of a large rule-based model for ERBB receptor signaling". Biologie systémů BMC. 6: 107. doi:10.1186/1752-0509-6-107. PMC 3485121. PMID 22913808.
- ^ A b Marchisio, M. A.; Colaiacovo, M; Whitehead, E; Stelling, J (2013). "Modular, rule-based modeling for the design of eukaryotic synthetic gene circuits". Biologie systémů BMC. 7: 42. doi:10.1186/1752-0509-7-42. PMC 3680069. PMID 23705868.
- ^ Chylek LA, Stites EC, Posner RG, Hlavacek WS (2013) Innovations of the rule-based modeling approach. In Systems Biology: Integrative Biology and Simulation Tools, Volume 1 (Prokop A, Csukás B, Editors), Springer.
- ^ Feret, J; Danos, V; Krivine, J; Harmer, R; Fontana, W (2009). "Internal coarse-graining of molecular systems". Sborník Národní akademie věd. 106 (16): 6453–8. Bibcode:2009PNAS..106.6453F. doi:10.1073/pnas.0809908106. PMC 2672529. PMID 19346467.
- ^ Dostupné v https://github.com/jkrivine/KaSim
- ^ A b C Hucka, M .; Finney, A .; Sauro, H. M .; Bolouri, H .; Doyle, J. C .; Kitano, H.; Arkin, A. P .; Bornstein, A. P .; Bray, B. J .; Cornish-Bowden, D .; Cuellar, A .; Dronov, A. A .; Gilles, S .; Ginkel, E. D .; Gor, M .; Goryanin, V .; Hedley, I. I .; Hodgman, W. J .; Hofmeyr, T. C .; Hunter, J. -H .; Juty, P. J .; Kasberger, N. S .; Kremling, J. L .; Kummer, A .; Le Novère, U .; Loew, N .; Lucio, L. M .; Mendes, P.; Minch, P .; Mjolsness, E. (2003). „Systémový biologický značkovací jazyk (SBML): médium pro reprezentaci a výměnu modelů biochemických sítí“. Bioinformatika. 19 (4): 524–531. doi:10.1093/bioinformatics/btg015. PMID 12611808.
- ^ Finney, A .; Hucka, M. (2003). „Značkovací jazyk biologie systémů: úroveň 2 a vyšší“ (PDF). Transakce s biochemickou společností. 31 (Pt 6): 1472–1473. CiteSeerX 10.1.1.466.8001. doi:10.1042 / bst0311472. PMID 14641091.[trvalý mrtvý odkaz ]
- ^ Monod, J .; Wyman, J .; Changeux, J. P. (1965). „O povaze alosterických přechodů: věrohodný model“. Journal of Molecular Biology. 12: 88–118. doi:10.1016 / S0022-2836 (65) 80285-6. PMID 14343300.
- ^ Colquhoun, D; Dowsland, K. A.; Beato, M; Plested, A. J. (2004). "How to impose microscopic reversibility in complex reaction mechanisms". Biofyzikální deník. 86 (6): 3510–8. Bibcode:2004BpJ....86.3510C. doi:10.1529/biophysj.103.038679. PMC 1304255. PMID 15189850.
- ^ A b John, M., Lhoussaine, C., Niehren, J., & Versari, C. (2011). Biochemical reaction rules with constraints. In Programming Languages and Systems (pp. 338-357). Springer Berlin Heidelberg.
- ^ Priami, C (1995). "Stochastic π-calculus". Počítačový deník. 38 (7): 578–589. doi:10.1093/comjnl/38.7.578.
- ^ A b C Gillespie, DT (1977). "Přesná stochastická simulace spojených chemických reakcí". J Phys Chem. 81 (25): 2340–2361. CiteSeerX 10.1.1.704.7634. doi:10.1021 / j100540a008.
- ^ A b C Maus, C; Rybacki, S; Uhrmacher, A. M. (2011). "Rule-based multi-level modeling of cell biological systems". Biologie systémů BMC. 5: 166. doi:10.1186/1752-0509-5-166. PMC 3306009. PMID 22005019.
- ^ A b J. Himmelspach and A. M. Uhrmacher, "Plug'n simulate," Proceedings of the 40th Annual Simulation Symposium. IEEE Computer Society, 2007, pp. 137-143.
- ^ Oury, N.; Plotkin, G. (2013). "Multi-level modelling via stochastic multi-level multiset rewriting" (PDF). Matematické struktury v informatice. 23 (2): 471–503. doi:10.1017/s0960129512000199.
- ^ A b Meier-Schellersheim, M; Xu, X; Angermann, B; Kunkel, E. J.; Jin, T; Germain, R. N. (2006). "Key role of local regulation in chemosensing revealed by a new molecular interaction-based modeling method". PLOS výpočetní biologie. 2 (7): e82. Bibcode:2006PLSCB...2...82M. doi:10.1371/journal.pcbi.0020082. PMC 1513273. PMID 16854213.
- ^ Angermann, B. R.; Klauschen, F; Garcia, A. D .; Prustel, T; Zhang, F; Germain, R. N.; Meier-Schellersheim, M (2012). "Computational modeling of cellular signaling processes embedded into dynamic spatial contexts". Přírodní metody. 9 (3): 283–9. doi:10.1038/nmeth.1861. PMC 3448286. PMID 22286385.
- ^ A b Lis, M; Artyomov, M. N.; Devadas, S; Chakraborty, A. K. (2009). "Efficient stochastic simulation of reaction-diffusion processes via direct compilation". Bioinformatika. 25 (17): 2289–91. doi:10.1093/bioinformatics/btp387. PMC 2734316. PMID 19578038.
- ^ Lopez, C. F.; Muhlich, J. L.; Bachman, J. A.; Sorger, P. K. (2013). "Programming biological models in Python using PySB". Molekulární systémy biologie. 9: 646. doi:10.1038/msb.2013.1. PMC 3588907. PMID 23423320.
- ^ Zhang F, Meier-Schellersheim M (2013) SBML Level 3 Package Specification: Multistate, Multicomponent and Multicompartment Species Package for SBML Level 3 (Multi). Version 1, Release 01 (Draft, Rev 369). Dostupné v http://sbml.org/Documents/Specifications/SBML_Level_3/Packages/multi
- ^ Tolle, DP; Nov, Le (2006). "Particle-Based Stochastic Simulation in Systems Biology". Curr. Bioinform. 1 (3): 315–320. doi:10.2174/157489306777827964. S2CID 41366617.
- ^ Michalski, P. J.; Loew, L. M. (2012). "CaMKII activation and dynamics are independent of the holoenzyme structure: An infinite subunit holoenzyme approximation". Physical Biology. 9 (3): 036010. Bibcode:2012PhBio...9c6010M. doi:10.1088/1478-3975/9/3/036010. PMC 3507550. PMID 22683827.
- ^ A b Mogilner, A; Allard, J; Wollman, R (2012). "Cell polarity: Quantitative modeling as a tool in cell biology". Věda. 336 (6078): 175–9. Bibcode:2012Sci...336..175M. doi:10.1126/science.1216380. PMID 22499937. S2CID 10491696.
- ^ Dostupné v http://sourceforge.net/projects/stochsim/
- ^ Levin, M. D.; Shimizu, T. S.; Bray, D (2002). "Binding and diffusion of CheR molecules within a cluster of membrane receptors". Biofyzikální deník. 82 (4): 1809–17. Bibcode:2002BpJ....82.1809L. doi:10.1016/S0006-3495(02)75531-8. PMC 1301978. PMID 11916840.
- ^ Yang, J; Hlavacek, W. S. (2011). "The efficiency of reactant site sampling in network-free simulation of rule-based models for biochemical systems". Physical Biology. 8 (5): 055009. Bibcode:2011PhBio...8e5009Y. doi:10.1088/1478-3975/8/5/055009. PMC 3168694. PMID 21832806.
- ^ A b Kinney, J. P.; Spacek, J; Bartol, T. M.; Bajaj, C. L.; Harris, K. M.; Sejnowski, T. J. (2013). "Extracellular sheets and tunnels modulate glutamate diffusion in hippocampal neuropil". Journal of Comparative Neurology. 521 (2): 448–64. doi:10.1002/cne.23181. PMC 3540825. PMID 22740128.
- ^ Plimpton S (1995) Fast Parallel Algorithms for Short-Range Molecular Dynamics. J Comput Phys 117:1-19
- ^ Dostupné v http://lammps.sandia.gov
- ^ Tschernyschkow, S; Herda, S; Gruenert, G; Döring, V; Görlich, D; Hofmeister, A; Hoischen, C; Dittrich, P; Diekmann, S; Ibrahim, B (2013). "Rule-based modeling and simulations of the inner kinetochore structure". Pokrok v biofyzice a molekulární biologii. 113 (1): 33–45. doi:10.1016/j.pbiomolbio.2013.03.010. PMID 23562479.
- ^ Dostupné v http://www.mcell.org
- ^ Dostupné v http://www.blender.org
- ^ Shimizu, T. S.; Aksenov, S. V.; Bray, D (2003). "A spatially extended stochastic model of the bacterial chemotaxis signalling pathway". Journal of Molecular Biology. 329 (2): 291–309. doi:10.1016/s0022-2836(03)00437-6. PMID 12758077.
- ^ Mirschel, S; Steinmetz, K; Rempel, M; Ginkel, M; Gilles, E. D. (2009). "PROMOT: Modular modeling for systems biology". Bioinformatika. 25 (5): 687–9. doi:10.1093/bioinformatics/btp029. PMC 2647835. PMID 19147665.
- ^ Hoops, S .; Sahle, S .; Gauges, R .; Lee, C .; Pahle, J.; Simus, N.; Singhal, M.; Xu, L .; Mendes, P .; Kummer, U. (2006). "COPASI--a COmplex PAthway SImulator". Bioinformatika. 22 (24): 3067–3074. doi:10.1093/bioinformatics/btl485. PMID 17032683.
- ^ Aitken, S; Alexander, R. D .; Beggs, J. D. (2013). "A rule-based kinetic model of RNA polymerase II C-terminal domain phosphorylation". Journal of the Royal Society Interface. 10 (86): 20130438. doi:10.1098/rsif.2013.0438. PMC 3730697. PMID 23804443.
- ^ Artyomov, M. N.; Lis, M; Devadas, S; Davis, M. M .; Chakraborty, A. K. (2010). "CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery". Sborník Národní akademie věd. 107 (39): 16916–21. Bibcode:2010PNAS..10716916A. doi:10.1073/pnas.1010568107. PMC 2947881. PMID 20837541.
- ^ Ibrahim, B., Henze, R., Gruenert, G., Egbert, M., Huwald, J., & Dittrich, P. (2013) Spatial Rule-Based Modeling: A Method and Its Application to the Human Mitotic Kinetochore. Cells (2073-4409), 2(3).