Waardenburgův syndrom typu 2D - Waardenburg Syndrome Type 2D
tento článek potřebuje víc lékařské odkazy pro ověření nebo se příliš spoléhá na primární zdroje. (Květen 2020) |
| Waardenburgův syndrom typu 2D | |
|---|---|
 | |
| Tato podmínka se dědí autozomálně recesivně | |
| Příčiny | mutace v genu SLUG (SNAI2) |
Waardenburgův syndrom typu 2D, podtyp Waardenburgův syndrom, je vzácná vrozená porucha způsobená mutací genu SLUG (SNAI2). Vyznačuje se nedostatkem pigmentace kůže, vlasů a očí, jakož i abnormalitami ve vnější stěně kochlea. Tento podtyp postrádá velkou vzdálenost mezi očima, známou jako dystopia canthorum, který je pozorován u většiny pacientů s Waardenburgovým syndromem. Postižení vykazují různé stupně hluchoty nebo úplné ztráty sluchu spolu s heterochromie a zprávy o časném šedivění. Toto onemocnění je pozorováno u novorozenec stadia raného života. [1].
Prezentace
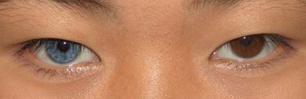
Typ 2 Waardenburgova syndromu je definován abnormalitami sluchové funkce a pigmentace ve vlasech, pokožce a očích a v některých případech progresivní ztrátou sluchu. Výsledkem je také heterochromie, kde oči mají různé barvy, asi u poloviny osob postižených typem 2.[2] Typ 2 také obsahuje variace možných genetických příčin, z nichž některé jsou způsobeny mutací v mikroftalmie asociovaný transkripční faktor (také známý jako MITF ) stejně jako SOX10 geny[3]. Typ 2D se odlišuje od ostatních typů absencí dystopia canthorum, což znamená, že u pacientů s typem 2D není vidět velká vzdálenost mezi očními dírkami přítomnými u jiných typů. A je to jediný podtyp Waardenburgova syndromu s autozomálně recesivní dědičností.[Citace je zapotřebí ]
Genetika
Typ 2D se dědí v autosomálně recesivní způsobem, proto oba rodiče postižených nesou recesivní gen. Ti, kteří nesou jednu kopii genu, nevykazují žádné příznaky onemocnění[4].
Bylo konkrétně zjištěno, že typ 2D je způsoben mutací genu SLUG, což je také protein se zinkovým prstem SNAI2. Gen SNAI2 je umístěn na chromozomu 8q11 a je tvořen třemi různými exony. Gen je zodpovědný za pigmentaci u myší, která je v souladu s lidským homologem. Myši bez přítomného genu SLUG vykazovaly kromě abnormalit pigmentace také dysfunkci v nervovém hřebenu [1][5]. Produkty genu jsou během vývoje aktivní v migračních buňkách v nervovém hřebenu. K dispozici je nejen tato funkce, ale také se prokázalo, že interakce s MITF gen, který je zodpovědný za různé typy Waardenburgova syndromu[1].
SNAI2 hraje hlavní roli v migraci buněk neurální lišty, ale nepodílí se na jejich tvorbě. Je také exprimován v několika částech těla během raného vývoje, včetně chondrocytů, mezenchymálních částí v plicích, vnitřnostech a ledvinách[5].
Delece tohoto SNAI2 byla studována u dvou velmi různorodých pacientů. Pacienti studovaní pro podtyp 2D byli oba vyšetřováni a vykazovali deleci obou kopií genu SLUG, což svědčí o tom, že tyto mutace vykazují příznaky u Waardenburgova syndromu typu 2D [1].
Diagnóza
Obecně má Waardenburgův syndrom pět hlavních kritérií, která jsou následující: vrozená ztráta sluchu, abnormalita pigmentace duhovky, abnormalita pigmentace vlasů, dystopia canthorum a příbuzný s Waardenburgovým syndromem. Jak již bylo zmíněno dříve, typ 2D nevykazuje žádné známky dystopia canthorum. Proto, aby mohla být jedinci diagnostikována dvě z hlavních kritérií vedle dystopia canthorum, musí vykazovat další hlavní příznaky. Často existuje nesprávná diagnóza mezi typem 1 a typem 2 Waardenburgova syndromu kvůli takovým blízkým podobnostem mezi pozorovaným fenotypem. Existuje také několik drobných znaků leukoderma, synophrys, široký nebo vysoký nosní můstek a hypoplázie nosních dír [6].
Řízení
V současné době neexistuje léčba Waardenburgova syndromu, doporučuje se však navštívit poradce, aby se přizpůsobili každodennímu životu. Ztráta sluchu může pro pacienty představovat určité potíže. Jiné příznaky, které se mohou objevit u pacientů se syndromem, uvedené jako vedlejší kritéria, mohou být léčeny, jako je hyperplazie a leucoderma, a měly by být léčeny podle potřeby.[Citace je zapotřebí ]
Dějiny
Dva pacienti byli studováni na typ 2D, kteří sdíleli stejnou genetickou mutaci v genu SLUG. Pacienti z různých prostředí, prvním pacientem je 15letá bangladéšská žena a druhým 3letým holandským chlapcem. První pacientka vykazovala ztrátu sluchu a žádné další příznaky a zbytek její rodiny nehlásil žádné podobné příznaky. Druhý pacient také hlásil ztrátu sluchu a heterochromii. Rodinná anamnéza ukázala, že mutace neměla vliv na žádné další členy[1]. Oba tito pacienti měli delece v genu SLUG, také v genu SNAI2 a MIFT, z nichž všichni hrají hlavní roli v časném vývoji neurální lišty.[Citace je zapotřebí ]
Reference
- ^ A b C d E Sánchez-Martín, Manuel; Rodríguez-García, Arancha; Pérez-Losada, Jesús; Sagrera, Ana; Přečtěte si, Andrew P .; Sánchez-García, Isidro (01.12.2002). „Delece SLUG (SNAI2) u pacientů s Waardenburgovou chorobou“. Lidská molekulární genetika. 11 (25): 3231–3236. doi:10,1093 / hmg / 11,25,3231. PMID 12444107.
- ^ Zhang, Hua; Luo, Hunjin; Chen, Hongsheng; Mei, Lingyun; On, Chufeng; Jiang, Lu; Li, Jia-Da; Feng, Yong (2012-11-30). "Funkční analýza mutací genu MITF spojených s Waardenburgovým syndromem typu 2". FEBS Dopisy. 586 (23): 4126–4131. doi:10.1016 / j.febslet.2012.10.006. ISSN 1873-3468. PMID 23098757. S2CID 33482135.
- ^ Baral, Viviane; Chaoui, Asma; Watanabe, Yuli; Goossens, Michel; Attie-Bitach, Tania; Marlin, Sandrine; Pingault, Veronique; Bondurand, Nadege (2012-07-27). „Screening MITF and SOX10 Regulatory Regions in Waardenburg Syndrome Type 2“. PLOS ONE. 7 (7): e41927. Bibcode:2012PLoSO ... 741927B. doi:10.1371 / journal.pone.0041927. ISSN 1932-6203. PMC 3407046. PMID 22848661.
- ^ Reference, Genetics Home. "Waardenburgův syndrom". Genetická domácí reference. Citováno 2020-04-30.
- ^ A b Pingault, Véronique; Ente, Dorothée; Dastot-Le Moal, Florencie; Goossens, Michel; Marlin, Sandrine; Bondurand, Nadège (duben 2010). "Recenze a aktualizace mutací způsobujících Waardenburgův syndrom". Lidská mutace. 31 (4): 391–406. doi:10,1002 / humu.21211. ISSN 1098-1004. PMID 20127975.
- ^ "Waardenburgův syndrom typu 2 | Informační centrum o genetických a vzácných onemocněních (GARD) - program NCATS". rarediseases.info.nih.gov. Citováno 2020-04-30.