Aza-Cope přesmyk - Aza-Cope rearrangement - Wikipedia
Přeskupení, zejména ta, kterých se může účastnit kaskádové reakce, jako aza-Cope přesmyky, mají v praxi velký praktický i koncepční význam organická chemie, kvůli jejich schopnosti rychle vybudovat strukturní složitost z jednoduchých výchozích materiálů. Aza-Cope přesmyky jsou příklady heteroatomových verzí Vyrovnejte přeskupení, což je [3,3] -sigmatropní přesmyk který posouvá jednoduché a dvojné vazby mezi dvěma allylický komponenty. V souladu s pravidly Woodward-Hoffmana probíhají termální aza-Cope přesmyky suprafaciálně.[1] Přesmyky Aza-Cope jsou obecně klasifikovány podle polohy dusíku v molekule (viz obrázek):

První příklad přesmyku aza-Cope byl všudypřítomný kationtové přeskupení 2-aza-Cope, ke kterému dochází při teplotách o 100–200 ° C nižších, než je přeskupení Cope, kvůli lehké povaze přesmyku.[2] Snadná povaha tohoto přesmyku je přičítána jednak skutečnosti, že kationtový 2-aza-Cope je ze své podstaty termoneutrální, což znamená, že neexistuje žádný předpětí pro výchozí materiál nebo produkt, jakož i přítomnost nabitý heteroatom v molekule, který snižuje aktivační bariéru. Méně časté jsou přeskupení 1-aza-Cope a přeskupení 3-aza-Cope, které jsou vzájemně mikroskopické. Přesmyky 1- a 3-aza-Cope mají vysoké aktivační bariéry a omezenou syntetickou použitelnost, což odpovídá jejich relativní nejasnosti.[3][4][5]
Aby se maximalizovala jeho syntetická využitelnost, je kationtový přesmyk 2-aza-Cope obvykle spárován s termodynamickým předpětím směrem k jedné straně přesmyku. Nejběžnější a synteticky nejvhodnější strategie spojuje kationtový 2-aza-Cope přesmyk s Mannichovou cyklizací, a je předmětem velké části tohoto článku. Tato tandemová aza-Cope / Mannichova reakce se vyznačuje mírnými reakčními podmínkami, diastereoselektivitou a širokou syntetickou použitelností. Poskytuje snadný přístup k substituovaným acylem pyrrolidiny, struktura běžně se vyskytující v přírodních produktech, jako je alkaloidy, a byl použit při syntéze řady z nich, zejména strychninu a krininu.[6] Larry E. Overman a spolupracovníci provedli rozsáhlý výzkum této reakce.[1]
Kationtový přesmyk 2-aza-Cope

Kationtový přesmyk 2-aza-Cope, který se nejlépe nazývá 2-azonia- [3,3] -sigmatropní přesmyk, byl důkladně studován Larry E. Overmanem a spolupracovníky. Jedná se o nejrozsáhlejší studii aza-Copeových přesmyků kvůli mírným podmínkám potřebným k provedení uspořádání a také kvůli mnoha syntetickým aplikacím, zejména při syntéze alkaloidů. Termodynamicky, obecný 2-aza-Cope přesmyk nemá zkreslení produktu, protože vazby rozbité a vytvořené jsou ekvivalentní v obou směrech reakce, podobně jako přeskupení Cope. Přítomnost iontového dusíkatého heteroatomu odpovídá za snadnější přeskupení kationtového přesmyku 2-aza-Cope ve srovnání s Copeovým přeskupením. Proto je často spárován s a termodynamický dřez ovlivnit produkt přeskupení.[1]
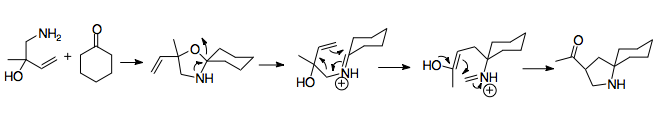
V roce 1950 uvedli Horowitz a Geissman první příklad přesmyku 2-aza-Cope, což byl překvapivý výsledek neúspěšného pokusu o syntézu aminoalkohol.[2] Tento objev identifikoval základní mechanismus přesmyku, protože produkt byl s největší pravděpodobností vyroben prostřednictvím dusíkatého analogu přesmyku Cope. Zpracování allylbenzylaminu (A) kyselinou mravenčí a formaldehydem vede k aminoalkoholu (B). Aminoalkohol se převádí na imin přidáním kyseliny (C), která prochází kationtovým přesmykem 2-aza-Cope (D). Voda hydrolyzuje iminiový ion na amin (E). Ošetření tohoto výchozího materiálu pouze formaldehydem ukázalo, že k alkylaci aminové skupiny došlo po kationtovém 2-aza-Cope přesmyku, což svědčí o rychlém zařízení přesmyku.[2]

Vzhledem k mírným zahřívacím podmínkám prováděné reakce, na rozdíl od těch přísnějších pro čistě uhlovodíkový přesmyk Cope, zavedl tento heteroatomový přesmyk Cope hypotézu, že pozitivní náboj na dusík v přeuspořádání cope významně snižuje aktivační bariéru pro přeskupení.[2]
Reakční mechanismus
Rychlost zrychlení díky kladně nabitému dusíku

Přesmyky aza-Cope předpovídá Vládne Woodward-Hoffman postupovat suprafacially. Přestože se Overman a jeho spolupracovníci nikdy výslovně nestudovali, předpokládali, že stejně jako u oxy-Cope přesmyk nabitý atom narušuje sigmatropní přesmyk z čistě sladěného reakčního mechanismu (jak se očekávalo v Copeově přesmyku) na jeden s částečným diradikálním / dipolárním charakterem v důsledku delokalizace pozitivního náboje na allylový fragment, který oslabuje allylovou vazbu. To má za následek sníženou aktivační bariéru pro rozbití vazby. Kationtový aza-Copeův přesmyk tedy probíhá rychleji než více koordinované procesy, jako je přesmyk Cope.[6][7]
Přechodový stav a stereochemie
Kationtový přesmyk 2-aza-Cope se vyznačuje svou vysokou stereospecificitou, která vyplývá z jeho vysoké preference pro přechodový stav židle. Při zkoumání stereospecificity tohoto přesmyku Overman a spolupracovníci použili logiku podobnou klasickým Doeringovým a Rothovým experimentům,[8] což ukázalo, že Copeův přeskupení preferuje konformaci židle.[9] Použitím kationtové reakce 2-aza-Cope / Mannich na prekurzorech pyrrolizidinu prokázali, že pyrrolizidiny s cis substituenty z E-alkenů a trans substituenty ze Z-alkenů jsou silně upřednostňovány, což svědčí o přechodném stavu křesla. Pokud by byl přechodový stav lodi funkční, byly by získány opačné výsledky (podrobně na obrázku níže).[9] Jak je u mnoha reakcí trendem, konverze Z-enolátu poskytuje nižší selektivitu díky 1,3 diaxiální sterické interakci mezi enolátem a kruhem, jakož i skutečnosti, že substituenty preferují kvaziekvatoriální umístění. To pomáhá vysvětlit vyšší teploty potřebné pro konverzi Z-enolátu.[6][9]Přechodový stav lodi je ještě méně upřednostňován přesmykem kationtového 2-aza-Cope, než je tomu u přesmyku Cope: v analogických situacích, kdy přesmyk Cope přebírá přechodový stav lodi, aza-Cope přesmyk pokračuje v křesle geometrie.[1][6][10] Tyto výsledky jsou v souladu s výsledky výpočetní chemie, které dále tvrdí, že přechodový stav je pod kinetickou kontrolou.[11]

Je příznačné, že tyto stereochemické experimenty naznačují, že kationtové přesmyky 2-aza-Cope (stejně jako Mannichova cyklizace) probíhají rychleji než tautomerizace enolem nebo iminiem. Pokud by tomu tak nebylo, nebyla by pozorována žádná smysluplná stereochemie, což by zdůrazňovalo možnosti této rychlé reakce.[1]
Další úvahy o stereochemii
Aza-Cope / Mannichova reakce, když se účastníte prstencové rozpínání, následuje stereochemii diktovanou nejpříznivější konformací křesla, která obecně umisťuje objemné substituenty kvazi-ekvatoriálně. Vinylové a aminové komponenty mohou mít po instalaci na prsten buď syn nebo anti vztah. Tento vztah je obvykle diktován aminovým substituentem: objemné substituenty vedou k prekurzorům syn aza-Cope. Zatímco proti vinylové a aminové substituenty mají obecně pouze jeden zvýhodněný přechodový stav, což vede k cis kondenzovanému kruhovému systému, výhodný produkt syn substituentů se může měnit, což je diktováno stérickými interakcemi s rozpouštědly nebo velkými N-substituenty, které mohou upřednostňovat před objemnými substituenty a měnit přechodový stav.[12][13]
U jednoduchých aza-Cope / Mannichových reakcí, které se neúčastní prstencové expanze, konkrétně kondenzace aminoalkoholů a etherů, dochází k rotaci vazby rychleji než Mannichova cyklizace a jsou pozorovány racemické produkty.[14] Tomu lze zabránit použitím chirálního pomocného substituentu na aminu. Reakce vázané na prsteny nemohou tyto vazby rotovat.[1]

Možné termodynamické výlevky pro předpětí produktu přesmyku
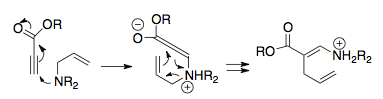
První příklad Horowitze a Geissmana demonstruje možné termodynamické jímky, které se spojí s kationtovým přesmykem 2-aza-Cope, kde je produkt předpjatý fenylovým substituentem prostřednictvím arylové konjugace, poté zachycen hydrolýzou iminia. Mezi další metody předpětí produktu patří použití substituentů, které jsou stabilnější na substituovaných uhlících, uvolnění kmene kruhu (například spárováním přesmyku s otvorem pro cyklopropan), intramolekulární zachycení (na obrázku) a spárování přeskupení s Mannichovou cyklizací.[1][15]

Aza-Cope / Mannichova reakce
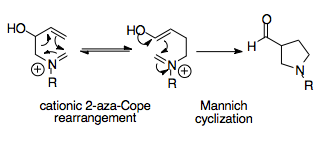
Reakce aza-Cope / Mannich je synteticky silná reakce, protože je schopna vytvářet složité cyklické molekuly z jednoduchých výchozích materiálů. Tato tandemová reakce poskytuje termodynamické předpětí vůči jednomu přesmyku produktu, protože Mannichova cyklizace je nevratná a její produkt, substituovaný acylem pyrrolidin prsten, stabilnější než přeskupení.[1][16]
První aza-Cope / Mannichova reakce
Overman a spolupracovníci si uvědomili, že kationtové přeskupení 2-aza-Cope může být potenciálně synteticky silné, pokud by bylo možné zavést vhodný termodynamický dřez. Jejich logikou bylo začlenit nukleofilní substituent do výchozího materiálu, konkrétně alkoholovou skupinu, která působí až po přeskupení, přeměněném na enol připraven k útoku na iminiový iont.
Tato první zpráva o reakci byla reakcí mezi aldehydy a 2-alkoxy-3-butenaminy, které vytvořily aminoalkohol, jehož produktem reakce aza-Cope / Mannich byl acyl-substituovaný pyrrolidinový kruh. Tento jednoduchý postup zahrnoval pouze mírné zahřívání po dobu několika hodin. Je příznačné, že reakce aza-Cope / Mannich probíhá v jednom kroku s vynikajícím výtěžkem. Tento postup lze snadno aplikovat na kondenzaci aminoetherů (viz níže), kde byl alkohol nejprve methylován.[16] Po provedení reakce aza-Cope / Mannich se keton vytvoří přidáním NaOH.[16] Amin, v tomto jednoduchém případě, nemůže tvořit iminiový ion ze základních ketonů; následující metody našel způsoby zabudování ketonů do reakce.[16][17] Užitečnost této reakce je evidentní ve skutečnosti, že i když se vytvoří méně stabilní izomer, reakce pokračuje, což prokazuje jeho vysokou termodynamickou výhodnost.[12][17]

Reakční mechanismus

Obecný produkt reakce může potenciálně nastat dvěma samostatnými cestami: aza-Cope / Mannichova reakce nebo aza-Prinsova cyklizace /pinacol přesmyk. Tyto mechanismy mají různé stereochemické vlastnosti, které objasňují dominanci aza-Cope / Mannichovy reakce. Reakce aza-Cope / Mannich donutí každý atom v analogu [1,5] dienu podstoupit sp2 hybridizace, vymazání stereochemie výchozího materiálu ve značené poloze R ', zatímco přesmyk aza-Prins / pinakol zachovává stereochemii ve značené poloze R', což ukazuje na jednoduchý test, který odhaluje aktivní mechanismus. Enantiomerně čistý výchozí materiál v poloze „R“ by měl vést k racemickému produktu, pokud je dominantním mechanismem reakce aza-Cope / Mannich, zatímco stereochemie by měla být zachována, pokud je dominantním mechanismem přesmyk cyklizace aza-prins / pinakol cesta. Jednoduchý experiment ověřil, že produkt byl racemický, a poskytl jasný důkaz o aza-Cope Mannichově reakci jako funkčním mechanismu. Další experimenty to ověřily s využitím znalostí, že karbeniový iont vytvořený v dráze aza-Prins / pinakol by byl ovlivněn schopností jeho substituentu stabilizovat jeho pozitivní náboj, čímž by se změnila reaktivita dráhy. Ukázalo se však, že řada substituentů má malý účinek na výsledek reakce, což opět ukazuje na Manzaovu reakci aza-Cope jako na operativní mechanismus.[14]Nedávná literatura z laboratoře Shanahan podporuje vzácnou cestu aza-Prins / pinakol spojenou pouze s významně zvýšenou nukleogenitou alkenu a elektrofilitou iminia.[1][6][18][19]
Reakce aza-Cope / Mannich vykazuje vysokou diastereoselektivitu, obecně v souladu s výsledky stereochemické experimenty objasňující přechodový stav kationtového přesmyku 2-aza-Cope, který následuje, protože tato tandemová reakční cesta byla nedílnou součástí těchto experimentů. Stereochemie přesmyku je o něco složitější, když allylové a aminové substituenty jsou instalovány na kruhu, a tedy navzájem cis nebo trans.
Výchozí materiál pro aza-Cope / Mannichovu reakci, aminoalkohol, lze také považovat za související s oxy-Cope přesmyk (níže), a to jak pro zrychlení rychlosti v důsledku iontového zapojení, tak pro analogickou funkci kolapsu enolu Mannichovy cyklizace a keto-enol tautomerizace v přeskupení oxy-Cope.[7]

Syntetické aplikace reakce 2-aza-Cope / Mannich
Aza-Cope / Mannichova reakce je často nejúčinnějším způsobem syntézy pyrrolidin kroužky, a má tedy řadu aplikací v totálních syntézách přírodních produktů. Vzhledem ke své diastereoselektivitě tato reakce přidala do katalogu nástrojů asymetrické syntézy, jak je vidět v mnoha příkladech asymetrické alkaloidy syntetizovány pomocí reakce. Jak jsme viděli v první aza-Cope / Mannichova reakce a při objasnění reakce stereochemie lze k vytvoření použít aza-Cope / Mannichovu reakci pyrrolidin prsteny a pyrrolizidin prsteny. Může být použit k vytvoření mnoha dalších kruhových struktur užitečných při syntéze, jako je indolizidin cykly a indol prsteny.[1][7]
Celková syntéza (-) - strychnin
Klasickým příkladem demonstrujícím užitečnost této reakce je Overmanova syntéza strychninu. Strychnin je přirozeně se vyskytující vysoce jedovatý alkaloid, nalezený v rodu stromů a lezení keřů Strychnos. Strychnin se běžně používá jako malý pesticid obratlovců. První celková syntéza strychninu autor: R. B. Woodward,[20] představoval hlavní krok v syntéze přírodních produktů: žádná molekula blížící se její složitosti nebyla dříve syntetizována. Další celkové syntézy byly hlášeny až koncem 80. let 20. století za použití podobných metod, konkrétně s použitím meziproduktu dostupného degradovaným strychninem. Všechny tyto syntézy používaly drsné podmínky. Overmanova syntéza obchází tyto problémy a je první asymetrickou celkovou syntézou strychninu, využívající výhod diastereoselektivity a mírných reakčních podmínek reakce aza-Cope / Mannich. Krok reakce aza-Cope / Mannich pokračoval v téměř kvantitativním výtěžku. Syntéza Overmana je tedy o několik řádů účinnější než její předchůdci.[20]

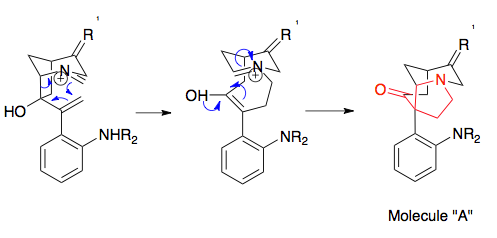
Overmanova syntéza strychninu představuje užitečný příklad přípravy prekurzorů nezbytných pro aza-Cope / Mannichův přesmyk, což představuje efektivní využití otevření epoxidového kruhu. Mezi klíčové kroky syntézy přeskupeného substrátu vedoucí k výchozím materiálům nezbytným pro reakci aza-Cope / Mannich patřila Stilleova reakce na sloučení dvou prekurzorů, epoxidace dvojné vazby pomocí terc-butylhydroperoxid, a Wittigova reakce převést keton na alken a krok cyklizace. Alkylace aminu (není zobrazena), transformuje molekulu na přesmykový substrát. Je příznačné, že tato molekula ukazuje enantiomerní přesnost reakce aza-Cope / Mannich, protože jednoduchý enantiomerní výchozí materiál určuje konečný enantiomer: enantiomer strychninu byl vyroben použitím enantiomeru výchozího materiálu.[20][21]

Syntéza Overmana s podrobnými podrobnostmi syntézy přeskupeného substrátu a závěrečných kroků reakce je podrobně popsána zde: Overmanova syntéza (-) - strychninu.
Syntéza (-) - krininu
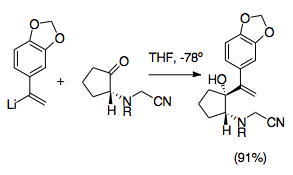
Crinine je alkaloid rodiny Amaryllidaceae a jeho asymetrická celková syntéza byla jednou z prvních využívajících aza-Cope / Mannichovu reakci. Tato syntéza představuje významný krok ve vývoji aza-Cope / Mannichovy reakce, protože využívá několik nejužitečnějších syntetických strategií charakteristických pro reakci. Tato reakce využívá vysokou diastereoselektivitu přeskupení kationtového 2-aza-Cope a také použití kyanomethylová skupina chránit amin během vinyllithium přidání a jako odstupující skupina pro podporu tvorby iminia, s pomocí přidání dusičnan stříbrný.[22]Tato syntéza je jedním příkladem mnoha z kyanomethylová skupina poskytnutí synteticky užitečné cesty k tvorbě pyrrolidinu a indolizidinu.

Syntéza přemostěných tricyklických alkaloidů
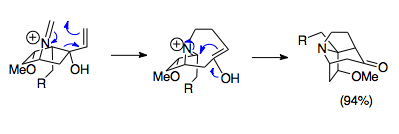
Overman a spolupracovníci vyvinuli metody syntézy komplikovaných přemostěných tricyklických struktur pomocí reakce aza-Cope / Mannich. Tyto azatricyklické struktury se nacházejí v komplexu Stemona alkaloidové rodiny, stejně jako v potenciálních drogách, jako jsou některé imunosupresiva. Uvedený příklad je snadná reakce kombinující výchozí materiál 1-aza-bicyklo [2.2.1] heptanové soli s paraformaldehydem při 80 ° C za vzniku stěžejní azatricyklické struktury Stemona molekuly alkaloidů. I přes nepříznivé orbitální překrytí způsobené steriky tohoto kruhového systému obvykle probíhá reakce s výtěžkem 94%, což zdůrazňuje sílu této reakce i za nepříznivých podmínek.[23]
Obecné otevírání a rozšiřování kroužku

K vytvoření se často používá aza-Cope / Mannichova reakce ve spojení s existujícími cykly kruhů indolizidin cykly (pyrrolidin připojený k cyklohexanovému kruhu). Tento typický prsten anulace, kde je cyklopentanová část otevřena přesmykem a uzavřena Mannichovou cyklizací za vzniku šestičlenného kruhu připojeného k pyrrolidinovému kruhu, zatímco nejpopulárnější aza-Cope / Mannichova anulace není jediná. Je také možné syntetizovat sedmičlenné kruhové cykly, protože enol a iminiové ionty zůstávají v dostatečně těsné blízkosti, aby podstoupily Mannichovu cyklizaci.[22] Syntéza makrocyklů nebyla při použití této reakce hlášena kvůli nedostatku blízkosti mezi enolem a iminiem.[6]Jako přeskupovací substráty lze také použít vinyloxazolidiny. Toto přeskupení se nejprve vytvoří vinyloxazolidin z ataku na cyklohexanon aminobutenolem, který poté prochází aza-Cope / Mannichovou reakcí za použití tepla a kyseliny (Lewis nebo protický). Tento příklad se rozbije a poté vytvoří pětičlenný kruh. Složitější příklady připojují oxazolidin k jinému kruhu a představují další metody pro tvorbu indolizidinových cyklů.[24]
Rozsah reakce aza-Cope / Mannich
Reakce aza-Cope / Mannich má ve srovnání s jinými metodami řadu výhod. Jemné podmínky reakce se neshodují: lehké zahřívání, obvykle ne vyšší než 80 ° C, široká škála rozpouštědel a přidání 1 stechiometrického ekvivalentu kyseliny, obvykle Kafrsulfonová kyselina (CSA) nebo Lewisova kyselina. Jiné způsoby syntézy pyrrolidinu nemohou konkurovat stereospecifičnosti, široké aplikace ve strukturách obsahujících deriváty pyrrolidinu a velký rozsah možných výchozích materiálů. Reakce se projevuje vysoká diastereoselektivita, a je robustní, pokračuje i když čelí špatnému orbitálnímu překrytí v přechodovém stavu.[1]

Výhody reakce aza-Cope / Mannich motivovaly výzkum syntézy výchozích materiálů pro reakci, který se rozdělil do dvou hlavních kategorií: přidání aminu a tvorba iminia (červená) a instalace vinylového substituentu (modrý). Při reakci lze použít širokou škálu N-substituentů (R), alkyl a aryl, z nichž některé ovlivňují stereochemický výsledek reakce. Vinylové skupiny jsou obecně omezeny na ty, které jsou buď 1,1 nebo 1,2-disubstituované (vinyl se substituenty na R1a R.1, R.2 respektive), přičemž byla tolerována široká škála elektronické a sterické odrůdy.[1]
Přidání aminu a tvorba iminia
Otevření epoxidového kruhu
Kruhový kmen epoxidů poskytuje užitečnou metodiku pro instalaci aminové skupiny o dva atomy od alkoholové skupiny. Epoxid může být nejprve rozbit bromidovým nukleofilním útokem. Primární aminy, aromatické aminy nebo anilidy lithné lze také použít jako nukleofily. Po tomto kroku často následuje ochranná O-methylace, která probíhá snadno.

Pokud sterici umožňují útok pouze na příslušný uhlík (koncový uhlík na rozdíl od druhého uhlíku), je účinný přímý útok intramolekulárním dusíkem, jako je tomu v případě syntéza strychninu.[16][25]
Tvorba iminiových iontů
Nejběžnějším způsobem generování iminiového iontu z nainstalovaného aminu je přidání formaldehyd nebo paraformaldehyd, který prochází kyselinou katalyzován kondenzace za vzniku iminia. Overmanova syntéza strychninu typizuje tuto metodu.[6][25] Příležitostně se používají intramolekulární karbonyly.[9] Mezi další způsoby tvorby iminiových iontů patří použití kyanomethylové skupiny nebo pomocí oxazolidiny jako prekurzory karbonylu.
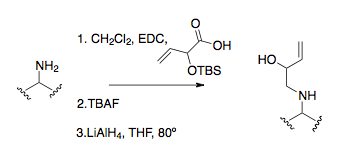
Alkylace aminu
Alkylace aminu představuje běžnou metodu pro získání iminových prekurzorů. Alkylace aminu přímým SN2 reakce je jen občas užitečná při výrobě výchozích materiálů kvůli vysoké náchylnosti aminů k nadalkylátu.[25]Redukční aminace je běžnější a účinnější alkylační postup, typické v prvním přesmyku aza-Cope.[16][26][27]Nejužitečnějším a standardním způsobem alkylace aminu je mít amin ve formě amidové vazby a následně ji redukovat, často LiAlH4.[9]
Použití oxazolidinu
Ketony a stéricky bráněné aldehydy nejsou vhodné pro základní aza-Cope / Mannichovu reakci, protože amin s nimi nemůže tvořit iminiový ion. Způsob, jak tento problém vyřešit, představuje tvorba dehydratovaného oxazolinu s následným zahříváním v přítomnosti plného ekvivalentu kyseliny. Společnost Overman uvádí použití oxizolidinů ke generování iminiového iontu nezbytného pro reakci. Po vytvoření Overman ukázal, že cyklohexanony mohou být použity pro karbonylovou složku při syntéze pyrrolidinu.[17] Tato reakce probíhala s různými formami cyklohexanonů. Když byl substituován acyklický keton, reakce probíhala s nízkým výtěžkem, což zvýrazňovalo termodynamickou výhodnost uvolňování cyklohexanonu z dvojně vázaného karbonylu, protože to vytváří nepříznivé vazebné napětí v konformaci křesla. To představuje jednu z nejvhodnějších konstrukcí 1-azaspiro [4,5] dekanového kruhového systému, což je užitečný přírodní produkt.[17]
Instalace vinylového substituentu
Vinylace ketonů
Vinylace může nabídnout další syntetické výhody umožňující rozšířenou funkčnost reakce.[23] Organolithná činidla se obvykle používají. Často se k dusíku přidá substituent nebo chránící skupina, i když to není vždy nutné. Přidání lithia k reakci má zásadní vliv na stereochemii výchozích materiálů, protože dusík k ní koordinuje. Výchozí materiály ovlivněné touto koordinací obecně vedou k prekurzorům anti aza-Cope, zatímco ty, které nejsou, jako jsou ty, které obsahují vysoce substituované, stéricky bráněné aminy, vedou k syn prekurzorům. Povaha dusíkatého substituentu má tedy velký význam.[6][25]
Použití kyanomethylové skupiny
Kyanomethylové skupiny představují snadný způsob ochrany iminiového iontu během alylické vinylace ketonu. Kyanamid skupiny a analogy se často používají při generování iminiových iontů. Obvykle se instalují nukleofilní adicí na iminiový iont, který se obvykle vyrábí alkylací aminu formaldehydem. Iminový iont je tak maskovaný.[28] Z toho vyplývá, že použití kyanomethylové skupiny poskytuje efektivní způsob řízení aza-Cope / Mannichovy reakce. Kyanomethylová skupina chrání dusík v poloze 2 během tvorby druhého allylického analogu logikou podobnou kyanidovému typu umpolung. Poté poskytuje dobrou odstupující skupinu pro tvorbu iminiového iontu v souladu s jeho použitím při generování iminiových iontů.[29] Generování iminiových iontů z kyanomethylových skupin je obvykle podporováno přidáním dusičnanu stříbrného, i když byly použity jiné sloučeniny stříbra a mědi. Tento přidaný krok umožňuje přesnější řízení tvorby iminiových iontů.[6][29] Důležité je, že tyto přípravné reakce musí být prováděny při -78 ° C, aby se zabránilo interakci kyanomethyl / vinyllithium. Tato metoda také umožňuje mnoho různých možných N-substituentů a lze ji použít ke zjednodušení syntézy oktahydroindolů a pyrroly.[1][29]

Přeuspořádání 1- a 3-aza-Cope

Přesmyky 1 a 3-aza-Cope jsou ve srovnání s kationtovým přesmykem 2-aza-Cope nejasné kvůli jejich aktivačním energiím, které jsou srovnatelně mnohem vyšší než u kationtového přesmyku 2-aza-Cope.
1- a 3-aza-Cope mají tendenci k tvorbě iminu na rozdíl od tvorby enaminu, protože π-vazba uhlík-dusík je silnější než σ-vazba uhlík-dusík, což znamená, že přesmyk 3-aza-Cope je termodynamicky upřednostňován, zatímco přeskupení 1-aza-Cope není: imin má téměř o 10 kcal / mol méně energie. Velké aktivační bariéry 3-aza Cope jsou tedy kineticky založeny.[30] Výzkum přeskupení 1 a 3-aza-Cope se zaměřil na nalezení dobrých hnacích sil ke snížení aktivačních bariér. Několik verzí těchto přeskupení bylo optimalizováno pro syntetické využití. Uspořádání 1-aza-Cope je obvykle spárováno s termodynamickými hnacími silami. 3-aza-Cope přesmyky se obecně provádějí kationicky, aby se snížila kinetická bariéra na termodynamicky příznivý produkt.[30][31]
Tato přeskupení následují po velké části mechanická logika kationtového přesmyku 2-Aza-Cope. Přeuspořádání 1 a 3-aza-Cope se obě přednostně vyskytují prostřednictvím přechodových stavů křesla (a zachovávají stereochemii, podobně jako kationtové přesmyky 2-aza-Cope), a jsou zrychlil se zavedením kladného náboje, protože to dává přechodovému stavu více diradical / dipolární charakter.[31] Očekává se, že přesmyk 3-aza-Cope (a tedy také přesmyk 1-aza-Cope, který prochází stejným přechodovým stavem) bude mít ve svém přechodovém stavu ještě méně aromatický charakter ve srovnání s přesmykem Cope a kationtovým 2- aza-Copeho přesmyk, který přispívá k vyšším požadovaným teplotám (blízkým teplotám požadovaným pro Copeův přesmyk, někdy ještě vyšším, od 170 do 300 stupňů) k překonání překážek kinetické aktivace těchto uspořádání.[3][31][32]
3-aza-Cope přesmyk

Reakce 3-aza-Cope byla objevena brzy po identifikaci přesmyku 2-aza-Cope kvůli analogickému vztahu k Claisenově přesmyku. Ve skutečnosti se v raných publikacích tato verze přesmyku aza-Cope často označuje jako přeskupení amino-Claisen, zkreslování přesmyku, protože by to znamenalo, že v molekule jsou dusík i kyslík.[3] Toto přeskupení lze použít k vytvoření heterocyklických kruhů zahrnujících uhlík, nejčastěji piperidin.
Jeden z prvních příkladů tohoto uspořádání identifikoval Burpitt, který poznal přesmyk vyskytující se v amonných solích, který vzhledem ke své nabité povaze probíhal exotermicky bez přidání tepla - což je důležité, bez přeskupeného dusíku přeskupení neproběhlo.[33] V návaznosti na tuto logiku se většina výzkumu přeskupení 3-aza-Cope zaměřila na nabité zwitteriontové verze této reakce, protože distribuce náboje pomáhá snižovat aktivační bariéru: v určitých případech může k přesmyku dojít při teplotách až - 20 ° C.[30][34]
HIll a Gilman poprvé ohlásili obecné nenabití 3-aza-Copeho přesmyku v roce 1967. Po vytvoření vhodně substituovaných enaminů poskytlo intenzivní zahřívání téměř úplné přeskupení iminového produktu. Tato cesta přeskupení má však omezenou užitečnost.[35]
1-aza-Cope přesmyk

První objevená reakce 1-aza-Cope byla jednoduchým analogem generické reakce Cope a vyžadovala intenzivní teplo k překonání své velké termodynamické aktivační bariéry; most subsequent work on the 1-aza-Cope rearrangement has thus focused on pairing the arrangement with a driving thermodynamic force to avoid these harsh reaction conditions. It has been hypothesized that the 1-aza-Cope rearrangement rate-determining transition state has partial diradical and dipolar transition state character due to the presence of the heteroatom.[4]
Fowler and coworkers have come up with a scheme that mobilizes the 1-aza-Cope rearrangement as a synthetically useful route.[3] Fowler and coworkers recognized that because the barrier for the reaction lies in the nitrogen's thermodynamic preference to stay as an imine, stabilizing the nitrogen could have a thermodynamically beneficial effect. To that end, Fowler and coworkers installed a carbonyl group on the nitrogen, hypothesizing that the lone pair of the nitrogen would be stabilized by participation in an amide bond, and that the electronegativity of this amide group would lower the LUMO of the imine group, making the transition state more favorable.[3] Using this strategy, Fowler and coworkers were able to use the 1-aza-Cope rearrangement to create piperidin a pyridin deriváty. This strategy was shown to be relatively robust, allowing for the formation of products even when forced through a boat transition state, when perturbed with substituent effects, or put in competition with alternative rearrangements.[3] Also significant is the relative ease of production of the reactants, which uses a Diels-Alder reaction paired with relatively simple workup steps, allowing for syntheses using complex cycling.[3]
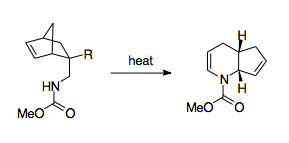
Other methods of overcoming this thermodynamic barrier include pairing it with cyclopropane ring strain release, which allows the reaction to proceed at much lower temperatures.[30][36]

Viz také
- Reviews by Overman[1][37] a Siegfried Blechert.[38]
Reference
- ^ A b C d E F G h i j k l m n Overman, L. E.; Humphreys, P. G.; Welmaker, G. S. (2011). "The Aza-Cope/Mannich Reaction". Organické reakce. 75. pp. 747–820. doi:10.1002/0471264180.or075.04. ISBN 978-0471264187.
- ^ A b C d Horowitz, R. M.; Geissman, T. A. (1950). "A Cleavage Reaction of α-Allylbenzylamines". J. Am. Chem. Soc. 72 (4): 1518–1522. doi:10.1021/ja01160a025.
- ^ A b C d E F G Chu M.; Wu P.L.; Givre S.; Fowler F.W. (1986). "The 1-AZA-Cope rearrangement". Čtyřstěn dopisy. 27 (4): 461–464. doi:10.1016/S0040-4039(00)85505-7.
- ^ A b Wu, P.L; Fowler, F. W. (1988). "The 1-aza-Cope rearrangement. 2". The Journal of Organic Chemistry. 53 (26): 5998–6005. doi:10.1021/jo00261a003.
- ^ Cook G.R.; Barta N.S.; Stille J.R. (1992). "Lewis acid-promoted 3-aza-Cope rearrangement of N-alkyl-N-allyl enamines". The Journal of Organic Chemistry. 57 (2): 461–467. doi:10.1021/jo00028a016.
- ^ A b C d E F G h i Overman, L.E.; Mendelson, L. T.; Jacobsen, E. J. (1983). "Synthesis applications of aza-Cope rearrangements. 12. Applications of cationic aza-Cope rearrangements for alkaloid synthesis. Stereoselective preparation of cis-3a-aryloctahydroindoles and a new short route to Amaryllidaceae alkaloids". J. Am. Chem. Soc. 105 (22): 6629–6637. doi:10.1021/ja00360a014.
- ^ A b C Overman, L. E. (1992). "Charge as a key component in reaction design. The invention of cationic cyclization reactions of importance in synthesis". Př. Chem. Res. 25 (8): 352–359. doi:10.1021/ar00020a005.
- ^ Doering, W.v.E.; Roth, W. R. (1962). "The overlap of two allyl radicals or a four-centered transition state in the cope rearrangement". Čtyřstěn. 18 (1): 67–74. doi:10.1016/0040-4020(62)80025-8.
- ^ A b C d E Doedens, R. J.; Meier, G.P.; Overman, L.E. (1988). "Synthesis applications of cationic aza-Cope rearrangements. Part 17. Transition-state geometry of [3,3]-sigmatropic rearrangements of iminium ions". J. Org. Chem. 53 (3): 685–690. doi:10.1021/jo00238a039.
- ^ Vogel, E.; Grimme, W.; Dinne, E. (December 1963). "Thermal Equilibrium between cis-1,2-Divinylcyclo-pentane and cis,cis-1,5-Cyclononadiene". Angewandte Chemie International Edition v angličtině. 2 (12): 739–740. doi:10.1002/anie.196307392.
- ^ Lukowski M.; Jacobs K.; Hsueh P.; Lindsay H.A; Milletti M.C. (2009). "Thermodynamic and kinetic factors in the aza-Cope rearrangement of a series of iminium cations". Čtyřstěn. 65 (50): 10311–10316. doi:10.1016/j.tet.2009.10.010.
- ^ A b McCann, S. F.; Overman, L. E. (1987). "Medium Effects and the Nature of the Rate-Determining Step in Mannich-Type Cyclizations". J. Am. Chem. Soc. 109 (20): 6107–6114. doi:10.1021/ja00254a033.
- ^ Overman, L. E.; Trenkle, W. C. (1997). "Controlling Stereoselection in Aza-Cope-Mannich Reactions". Isr. J. Chem. 37: 23–30. doi:10.1002/ijch.199700005.
- ^ A b Jacobsen E. J.; Levin J.; Overman L. E. (1988). "Synthesis applications of cationic aza-Cope rearrangements. Part 18. Scope and mechanism of tandem cationic aza-Cope rearrangement-Mannich cyclization reactions". J. Am. Chem. Soc. 110 (13): 4329–4336. doi:10.1021/ja00221a037.
- ^ Marshall, J. A .; Babler, J. H. (1969). "Heterolytic fragmentation of 1-substituted decahydroquinolines". J. Org. Chem. 34 (12): 4186–4188. doi:10.1021/jo01264a104.
- ^ A b C d E F Overman L. E.; Kakimoto, M. (1979). "Carbon-Carbon Bond Formation via Directed 2-Azonia-[3,3]-Sigmatropic Rearrangements. A New Pyrrolidine Synthesis". J. Am. Chem. Soc. 101 (5): 1310–1312. doi:10.1021/ja00499a058.
- ^ A b C d Overman L.E.; Kakimoto M.; Okawara M. (1979). "Directed 2-azonia-[3,3]-sigmatropic rearrangements. a convenient preparation of substituted 1-azaspiro[4,5]decanes". Čtyřstěn dopisy. 20 (42): 4041–4044. doi:10.1016/s0040-4039(01)86498-4.
- ^ Armstrong, A.; Shanahan, S. E. (2005). "aza-Prins-pinacol approach to 7-azabicyclo[2.2.1]heptanes and ring expansion to [3.2.1]tropanes". Org. Lett. 7: 1335. doi:10.1021/ja00221a037.
- ^ aza-Prins-pinacol approach to 7-azabicyclo[2.2.1]heptanes and ring expansion to [3.2.1]tropanes Armstrong, A.; Shanahan, S. E. Org. Lett. 2005, 7, 1335
- ^ A b C d R. B. Woodward; M. P. Cava; W. D. Ollis; A. Hunger; H. U. Daeniker; K. Schenker (1963). "The total synthesis of strychnine". Čtyřstěn. 19 (2): 247–288. doi:10.1016/S0040-4020(01)98529-1. PMID 13305562.
- ^ Knight, S.D.; Overman, L. E.; Pairaudeau, G. (1993). "Synthesis applications of cationic aza-Cope rearrangements. 26. Enantioselective total synthesis of (−)-strychnine". J. Am. Chem. Soc. 115 (20): 9293–9294. doi:10.1021 / ja00073a057.
- ^ A b Overman, L. E.; Sugai, s. (1985). "Total Synthesis of (−)-Crinine. Use of Tandem Cationic Aza-Cope Rearrangement/Mannich Cyclizations for the Synthesis of Enantiomerically Pure Amaryllidaceae Alkaloids". Helv. Chim. Acta. 68 (3): 745–749. doi:10.1002/hlca.19850680324.
- ^ A b Brueggemann, M.; McDonald, A. I.; Overman, L.E.; Rosen, M.D.; Schwink, L.; Scott, J.P. (2003). "Total Synthesis of (±)-Didehydrostemofoline (Asparagamine A) and (±)-Isodidehydrostemofoline". J. Am. Chem. Soc. 125 (50): 15284–15285. doi:10.1021/ja0388820. PMID 14664560.
- ^ Overman, L. E.; Shim, J. (1993). "Synthesis applications of cationic aza-Cope rearrangements. Part 25. Total synthesis of Amaryllidaceae alkaloids of the 5,11-methanomorphanthridine type. Efficient total syntheses of (−)-pancracine and (.+-.)-pancracine". Organické reakce. 58 (17): 4662–4672. doi:10.1021/jo00069a032.
- ^ A b C d Overman L. E.; Kakimoto, M.; Okazaki, M.E.; Meier, G.P. (1983). "Synthesis applications of aza-Cope rearrangements. 11. Carbon-carbon bond formation under mild conditions via tandem cationic aza-Cope rearrangement-Mannich reactions. A convenient synthesis of polysubstituted pyrrolidines". J. Am. Chem. Soc. 105 (22): 6622–6629. doi:10.1021/ja00360a013.
- ^ Overman, L.E.; Fukaya, C. (1980). "Stereoselective total synthesis of (.+-.)-perhydrogephyrotoxin. Synthetic applications of directed 2-azonia-[3,3]-sigmatropic rearrangements". J. Am. Chem. Soc. 102 (4): 1454–1456. doi:10.1021/ja00524a057.
- ^ Borch,R. F.; Bernstein, M. D.; Durst H. D. (1971). "Cyanohydridoborate anion as a selective reducing agent". J. Am. Chem. Soc. 93 (12): 2897–2904. doi:10.1021/ja00741a013.
- ^ Grierson D. S.; Harris, M .; Husson, H.P. (1980). "Synthesis and chemistry of 5,6-dihydropyridinium salt adducts. Synthons for general electrophilic and nucleophilic substitution of the piperidine ring system". J. Am. Chem. Soc. 102 (3): 1064–1082. doi:10.1021/ja00523a026.
- ^ A b C Overman, L. E.; Jacobsen, E. J. (1982). "The cyanomethyl group for nitrogen protection and iminium ion generation in ring-enlarging pyrrolidine annulations. A short synthesis of the amaryllidaceae alkaloid d,1-crinine". Tetrahedron Lett. 67 (51): 2741–2744. doi:10.1016/S0040-4039(00)87446-8.
- ^ A b C d http://www.chem.uky.edu/research/cammers/thesis-pdf/2.pdf
- ^ A b C Jolidon, S.; Hansen, H. J. (1997). "Untersuchungen über aromatische Amino-Claisen-Umlagerungen". Helv. Chim. Acta. 60 (2): 978–1032. doi:10.1002/hlca.19770600329.
- ^ Zahedi Ehsan; Ali-Asgari Safa; Keley Vahid (2010). "NBO and NICS analysis of the allylic rearrangements (the Cope and 3-aza-Cope rearrangements) of hexa-1,5-diene and N-vinylprop-2-en-1-amine: A DFT study". Central European Journal of Chemistry. 8 (5): 1097–1104. doi:10.2478/s11532-010-0084-1.
- ^ Brannock Kent; Burpitt Robert (1961). "Notes- The Chemistry of Isobutenylamines. II. Alkylation with Allylic and Benzyl Halides". J. Org. Chem. 26 (9): 3576–3577. doi:10.1021/jo01067a645.
- ^ Baxter, E. W.; Labaree, D.; Ammon, H. L.; Mariano, P. S. (1990). "Formal total synthesis of deserpidine demonstrating a versatile amino-Claisen rearrangement/Wenkert cyclization strategy for the preparation of functionalized yohimbane ring systems". J. Am. Chem. Soc. 12 (21): 7682–7692. doi:10.1021/ja00177a032.
- ^ Hill, R. K.; Gilman, N. W. (1967). "A nitrogen analog of the Claisen rearrangement". Čtyřstěn dopisy. 8 (15): 1421–1423. doi:10.1016/S0040-4039(00)71596-6.
- ^ Boeckman, R. K.; Shair, M.D.; Vargas, R. J.; Stolz, L. A. (1993). "Synthetic and Mechanistic Studies of the retro-Claisen Rearrangement. 2. A Facile route to Medium-Ring Heterocycles via Rearrangement of Vinylcyclopropane- and Cyclobutanecarboxaldehydes". J. Org. Chem. 58 (2): 1295–1297. doi:10.1021/jo00058a001.
- ^ Overman, L. E. (2009). "Molecular rearrangements in the construction of complex molecules". Čtyřstěn. 65 (33): 6432–6446. doi:10.1016/j.tet.2009.05.067. PMC 2902795. PMID 20640042.
- ^ Siegfried Blechert (1989). "The Hetero-Cope Rearrangement in Organic Synthesis". Syntéza. 1989 (2): 71–82. doi:10.1055/s-1989-27158.