Soused se připojil - Neighbor joining
v bioinformatika, soused se připojí je zdola nahoru (aglomerativní) shlukování metoda pro vytvoření fylogenetické stromy, vytvořil Naruya Saitou a Masatoshi Nei v roce 1987.[1] Obvykle se používá pro stromy založené na DNA nebo protein sekvence data, algoritmus vyžaduje znalost vzdálenosti mezi každou dvojicí taxony (např. druh nebo sekvence) k vytvoření stromu.[2]
Algoritmus

Připojení souseda se považuje za vstup a matice vzdálenosti určující vzdálenost mezi každou dvojicí taxonů. Algoritmus začíná zcela nevyřešeným stromem, jehož topologie odpovídá topologii hvězdná síť, a iteruje v následujících krocích, dokud není strom zcela vyřešen a jsou známy všechny délky větví:
- Na základě aktuální matice vzdálenosti vypočítejte matici (definováno níže).
- Najděte pár odlišných taxonů i a j (tj. S ) pro který má nejnižší hodnotu. Tyto taxony jsou spojeny s nově vytvořeným uzlem, který je připojen k centrálnímu uzlu. Na obrázku vpravo jsou f a g spojena s novým uzlem u.
- Vypočítejte vzdálenost od každého z taxony v páru k tomuto novému uzlu.
- Vypočítejte vzdálenost od každého z taxonů mimo tento pár k novému uzlu.
- Spusťte algoritmus znovu, nahraďte pár spojených sousedů novým uzlem a použijte vzdálenosti vypočítané v předchozím kroku.



Q-matice
Na základě matice vzdálenosti vztahující se k taxony, vypočítat jak následuje:

(1)

kde je vzdálenost mezi taxony a .



Vzdálenost od párových členů k novému uzlu
Pro každý z taxonů ve spojovaném páru použijte k výpočtu vzdálenosti k novému uzlu následující vzorec:
(2)
![delta (f, u) = { frac {1} {2}} d (f, g) + { frac {1} {2 (n-2)}} left [ sum _ {{k = 1}} ^ {n} d (f, k) - součet _ {{k = 1}} ^ {n} d (g, k) doprava] quad](https://wikimedia.org/api/rest_v1/media/math/render/svg/fbb5a4b2a6fbec48a4bbc6d6fc1e351e5eb851e0)
a:

Taxony a jsou spárované taxony a je nově vytvořený uzel. Spojení větví a a a a jejich délky, a jsou součástí stromu, který se postupně vytváří; neovlivňují ani nejsou ovlivněny pozdějšími kroky spojování sousedů.





Vzdálenost ostatních taxonů od nového uzlu
Pro každý taxon neuvažovaný v předchozím kroku vypočítáme vzdálenost k novému uzlu následujícím způsobem:
(3)
![d (u, k) = { frac {1} {2}} [d (f, k) + d (g, k) -d (f, g)]](https://wikimedia.org/api/rest_v1/media/math/render/svg/29d8e77a0543fb3258d08396a6fb44d387a92ad8)
kde je nový uzel, je uzel, do kterého chceme vypočítat vzdálenost do a a jsou členy právě připojeného páru.

Složitost
Soused se připojil k souboru taxa vyžaduje iterace. V každém kroku musí člověk vytvořit a hledat a matice. Zpočátku matice je velikost , pak je další krok Implementace tohoto přímým způsobem vede k algoritmu s časovou složitostí ;[3] existují implementace, které používají heuristiku k tomu, aby v průměru dosáhly mnohem lepších výsledků.[4]




Příklad
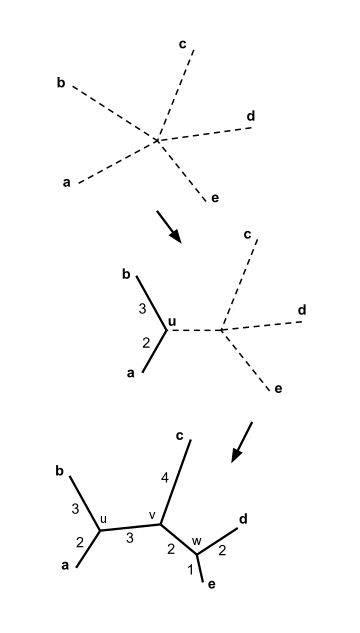
Předpokládejme, že máme pět taxonů a následující matice vzdálenosti :


| A | b | C | d | E | |
|---|---|---|---|---|---|
| A | 0 | 5 | 9 | 9 | 8 |
| b | 5 | 0 | 10 | 10 | 9 |
| C | 9 | 10 | 0 | 8 | 7 |
| d | 9 | 10 | 8 | 0 | 3 |
| E | 8 | 9 | 7 | 3 | 0 |
První krok
První připojení
Vypočítáme hodnoty rovnicí (1). Například:



Získáváme následující hodnoty pro matice (diagonální prvky matice se zde nepoužívají a jsou zde uvedeny):
| A | b | C | d | E | |
|---|---|---|---|---|---|
| A | −50 | −38 | −34 | −34 | |
| b | −50 | −38 | −34 | −34 | |
| C | −38 | −38 | −40 | −40 | |
| d | −34 | −34 | −40 | −48 | |
| E | −34 | −34 | −40 | −48 |
Ve výše uvedeném příkladu . Toto je nejmenší hodnota , takže spojujeme prvky a .



Odhad délky první větve
Nechat označit nový uzel. Rovnicí (2), nahoře, větve spojující a na pak mají délky:
![{ displaystyle delta (a, u) = { frac {1} {2}} d (a, b) + { frac {1} {2 (5-2)}} vlevo [ sum _ { k = 1} ^ {5} d (a, k) - sum _ {k = 1} ^ {5} d (b, k) right] quad = { frac {5} {2}} + { frac {31-34} {6}} = 2}](https://wikimedia.org/api/rest_v1/media/math/render/svg/37ab7ad46b3277cb0d8fa097d54007520aadb1df)

První aktualizace matice vzdálenosti
Poté pokračujeme v aktualizaci počáteční matice vzdálenosti do nové matice vzdálenosti (viz níže), zmenšené o jeden řádek a jeden sloupec kvůli spojení s do svého souseda . Pomocí rovnice (3) výše vypočítáme vzdálenost od do každého z ostatních uzlů kromě toho a . V tomto případě získáme:

![{ displaystyle d (u, c) = { frac {1} {2}} [d (a, c) + d (b, c) -d (a, b)] = { frac {9 + 10 -5} {2}} = 7}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7f4eedbd420cf866e0aca938b6b92ac75e7a332d)
![{ displaystyle d (u, d) = { frac {1} {2}} [d (a, d) + d (b, d) -d (a, b)] = { frac {9 + 10 -5} {2}} = 7}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4c547916160e8c48cdf86f8c07bf9dc4fb079aa1)
![{ displaystyle d (u, e) = { frac {1} {2}} [d (a, e) + d (b, e) -d (a, b)] = { frac {8 + 9 -5} {2}} = 6}](https://wikimedia.org/api/rest_v1/media/math/render/svg/498737ead33eb4e18634803a7c2a940091865cb1)
Výsledná matice vzdálenosti je:
| u | C | d | E | |
|---|---|---|---|---|
| u | 0 | 7 | 7 | 6 |
| C | 7 | 0 | 8 | 7 |
| d | 7 | 8 | 0 | 3 |
| E | 6 | 7 | 3 | 0 |
Tučné hodnoty v odpovídají nově vypočítaným vzdálenostem, zatímco kurzívou hodnoty nejsou aktualizací matice ovlivněny, protože odpovídají vzdálenostem mezi prvky, které nebyly zahrnuty do prvního spojení taxonů.
Druhý krok
Druhé připojení
Korespondence matice je:

| u | C | d | E | |
|---|---|---|---|---|
| u | −28 | −24 | −24 | |
| C | −28 | −24 | −24 | |
| d | −24 | −24 | −28 | |
| E | −24 | −24 | −28 |
Můžeme se rozhodnout buď se připojit a , nebo se připojit a ; oba páry mají minimum hodnota a obě volby vedou ke stejnému výsledku. Pro úplnost se připojme a a zavolat nový uzel .





Odhad délky druhé větve
Délky spojujících se větví a na lze vypočítat:
![{ displaystyle delta (u, v) = { frac {1} {2}} d (u, c) + { frac {1} {2 (4-2)}} vlevo [ sum _ { k = 1} ^ {4} d (u, k) - sum _ {k = 1} ^ {4} d (c, k) right] quad = { frac {7} {2}} + { frac {20-22} {4}} = 3}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a469c064887abf42cd02ed9383f0fabff3d50379)

Spojení prvků a výpočet délky větve pomáhají nakreslit sousední spojovací strom jak je znázorněno na obrázku.
Aktualizace druhé matice vzdálenosti
Aktualizovaná matice vzdálenosti pro zbývající 3 uzly, , , a , je nyní vypočítán:

![{ displaystyle d (v, d) = { frac {1} {2}} [d (u, d) + d (c, d) -d (u, c)] = { frac {7 + 8 -7} {2}} = 4}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9a149fca7dbf5339b8aaf177e55baf3ae19e3de6)
![{ displaystyle d (v, e) = { frac {1} {2}} [d (u, e) + d (c, e) -d (u, c)] = { frac {6 + 7 -7} {2}} = 3}](https://wikimedia.org/api/rest_v1/media/math/render/svg/75942d802678c69bac9b44e8b213994e490fe44c)
| proti | d | E | |
|---|---|---|---|
| proti | 0 | 4 | 3 |
| d | 4 | 0 | 3 |
| E | 3 | 3 | 0 |
Poslední krok
Stromová topologie je v tomto okamžiku plně vyřešena. Pro přehlednost však můžeme vypočítat matice. Například:


| proti | d | E | |
|---|---|---|---|
| proti | −10 | −10 | |
| d | −10 | −10 | |
| E | −10 | −10 |
Pro úplnost se připojme a a zavolat poslední uzel . Lze vypočítat délky tří zbývajících větví:

![{ displaystyle delta (v, w) = { frac {1} {2}} d (v, d) + { frac {1} {2 (3-2)}} vlevo [ sum _ { k = 1} ^ {3} d (v, k) - součet _ {k = 1} ^ {3} d (d, k) doprava] quad = { frac {4} {2}} + { frac {7-7} {2}} = 2}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7ef24fb03c3726d7607adbd331c3301b26e40551)


Sousední spojovací strom je nyní kompletní, jak je znázorněno na obrázku.
Závěr: aditivní vzdálenosti
Tento příklad představuje idealizovaný případ: všimněte si, že pokud se přesuneme z kteréhokoli taxonu na jakýkoli jiný podél větví stromu a sečteme délky procházejících větví, výsledek se rovná vzdálenosti mezi těmito taxony ve vstupní matici vzdálenosti. Například jít z na my máme . Distanční matice, jejíž vzdálenosti tímto způsobem souhlasí s nějakým stromem, se říká „aditivní“, což je v praxi vlastnost, která je vzácná. Je však důležité si uvědomit, že vzhledem k aditivní matici vzdálenosti jako vstupu je zaručeno, že sousední spojení najde strom, jehož vzdálenosti mezi taxony s ním souhlasí.

Soused se připojil jako minimální vývoj
Sousední připojení lze zobrazit jako a chamtivý heurista pro Vyvážená minimální evoluce[5] (BME) kritérium. Pro každou topologii definuje BME délku stromu (součet délek větví) jako konkrétní vážený součet vzdáleností v matici vzdáleností, přičemž váhy závisí na topologii. Optimální topologie BME je ta, která minimalizuje tuto délku stromu. Soused, který se připojuje na každém kroku, se chamtivě připojuje k této dvojici taxonů, což způsobí největší snížení odhadované délky stromu. Tento postup nezaručuje nalezení optima pro kritérium BME, i když to často dělá a je obvykle docela blízko.
Výhody a nevýhody
Hlavní ctností NJ je, že je rychlý[6]:466 ve srovnání s nejmenší čtverce, maximální šetrnost a maximální pravděpodobnost metody.[6]Díky tomu je praktické pro analýzu velkých souborů dat (stovky nebo tisíce taxonů) a pro bootstrapping, pro které účely se používají jiné analytické prostředky (např. maximální šetrnost, maximální pravděpodobnost ) možná výpočetně prohibitivní.
Sousední spojení má tu vlastnost, že pokud je vstupní matice vzdálenosti správná, pak bude výstupní strom správný. Dále je zaručena správnost topologie výstupního stromu, pokud je matice vzdálenosti „téměř aditivní“, konkrétně pokud je každá položka v matice vzdálenosti se liší od skutečné vzdálenosti o méně než polovinu nejkratší délky větve ve stromu.[7]V praxi distanční matice zřídka splňuje tuto podmínku, ale sousední spojení často stejně vytváří správnou stromovou topologii.[8] Správnost spojení sousedů pro téměř aditivní matice vzdálenosti znamená, že je statisticky konzistentní podle mnoha modelů evoluce; vzhledem k dostatečné délce dat, připojení sousedů rekonstruuje skutečný strom s vysokou pravděpodobností. ve srovnání s UPGMA a WPGMA, sousední spojení má tu výhodu, že nepředpokládá, že se všechny linie budou vyvíjet stejnou rychlostí (hypotéza molekulárních hodin ).
Přesto je sousedské připojení do značné míry nahrazeno fylogenetickými metodami, které se nespoléhají na měření vzdálenosti a za většiny podmínek nabízejí vynikající přesnost.[Citace je zapotřebí ] Sousední spojení má nežádoucí vlastnost, že některým z větví často přiřazuje záporné délky.
Implementace a varianty
Existuje mnoho programů implementujících připojení sousedů. RapidNJ aNINJA jsou rychlé implementace s typickými dobami chodu úměrnými přibližně druhé mocnině počtu taxonů.BIONJ a We Neighbor jsou varianty spojování sousedů, které zlepšují jeho přesnost využitím skutečnosti, že kratší vzdálenosti v matici vzdáleností jsou obecně lépe známy než delší vzdálenosti. FastME je implementace úzce související metody vyváženého minimálního vývoje.
Viz také
Reference
- ^ Saitou, N .; Nei, M. (1. července 1987). „Metoda spojování sousedů: nová metoda rekonstrukce fylogenetických stromů“. Molekulární biologie a evoluce. 4 (4): 406–425. doi:10.1093 / oxfordjournals.molbev.a040454. PMID 3447015.
- ^ Xavier Didelot (2010). „Sekvenční analýza struktur bakteriální populace“. V D. Ashley Robinson; Daniel Falush; Edward J. Feil (eds.). Genetika bakteriální populace u infekčních nemocí. John Wiley and Sons. s. 46–47. ISBN 978-0-470-42474-2.
- ^ Studier, J. A .; Keppler, K. J. (listopad 1988). „Poznámka k algoritmu Saitou a Nei pro spojování sousedů“. Molekulární biologie a evoluce. 5 (6): 729–31. doi:10.1093 / oxfordjournals.molbev.a040527. ISSN 1537-1719. PMID 3221794.
- ^ Mailund, Thomas; Brodal, GerthS; Fagerberg, Rolf; Pedersen, ChristianNS; Phillips, Derek (2006). „Přepracování metody spojování sousedů“. BMC bioinformatika. 7 (1): 29. doi:10.1186/1471-2105-7-29. PMC 3271233. PMID 16423304.
- ^ Gascuel O, Ocel M. (2006). „Sousední spojení odhaleno“. Mol Biol Evol. 23 (11): 1997–2000. doi:10.1093 / molbev / msl072. PMID 16877499.
- ^ A b Kuhner, M. K .; Felsenstein, J. (01.05.1994). „Simulační srovnání fylogenetických algoritmů za stejných a nerovných evolučních rychlostí“. Molekulární biologie a evoluce. 11 (3): 459–468. doi:10.1093 / oxfordjournals.molbev.a040126. ISSN 0737-4038. PMID 8015439.
- ^ Atteson K (1997). „Výkon algoritmů spojování sousedů při rekonstrukci fylogeneze“, str. 101–110. v Jiang, T. a Lee, D., eds., Přednášky z informatiky, 1276, Springer-Verlag, Berlín. COCOON '97.
- ^ Mihaescu R, Levy D, Pachter L. (2009). "Proč sousedské spojení funguje". Algorithmica. 54 (1): 1–24. arXiv:cs / 0602041. doi:10.1007 / s00453-007-9116-4. S2CID 2462145.CS1 maint: více jmen: seznam autorů (odkaz)
Jiné zdroje
- Studier JA, Keppler KJ (1988). „Poznámka k algoritmu Saitou a Nei spojujícího souseda“. Mol Biol Evol. 5 (6): 729–731. doi:10.1093 / oxfordjournals.molbev.a040527. PMID 3221794.
- Martin Simonsen; Thomas Mailund; Christian N. S. Pedersen (2008). Rychlé sousedské připojení (PDF). Sborník WABI. Přednášky z informatiky. 5251. str. 113–122. CiteSeerX 10.1.1.218.2078. doi:10.1007/978-3-540-87361-7_10. ISBN 978-3-540-87360-0.[trvalý mrtvý odkaz ]
externí odkazy
- Metoda sousedství - výukový program
| Relevantní pole | ||
|---|---|---|
| Základní pojmy | ||
| Inferenční metody | ||
| Aktuální témata | ||
| Skupinové vlastnosti | ||
| Skupinové typy | ||
| Nomenklatura | ||
| ||