UPGMA - UPGMA - Wikipedia
UPGMA (metoda neváženého páru s aritmetickým průměrem) je jednoduchá aglomerace (zdola nahoru) hierarchické shlukování metoda. Metoda je obecně přičítána Sokal a Michener.[1]
Metoda UPGMA je podobná metodě vážený varianta, WPGMA metoda.
Všimněte si, že nevážený člen naznačuje, že všechny vzdálenosti přispívají rovnoměrně ke každému průměru, který je počítán, a neodkazuje na matematiku, jíž je dosaženo. Jednoduché průměrování ve WPGMA tedy vytváří vážený výsledek a proporcionální průměrování v UPGMA vytváří nevážený výsledek (viz pracovní příklad ).[2]
Algoritmus
Algoritmus UPGMA vytváří zakořeněný strom (dendrogram ), která odráží strukturu přítomnou v páru matice podobnosti (nebo a matice odlišnosti V každém kroku se nejbližší dva klastry spojí do klastru vyšší úrovně. Vzdálenost mezi libovolnými dvěma klastry a , každá o velikosti (tj., mohutnost ) a , je považován za průměr všech vzdáleností mezi dvojicemi předmětů v a v , tj. Střední vzdálenost mezi prvky každého klastru:








Jinými slovy, v každém kroku vytváření klastrů aktualizovaná vzdálenost mezi spojenými klastry a nový klastr je dáno proporcionálním průměrováním a vzdálenosti:





Algoritmus UPGMA vytváří zakořeněné dendrogramy a vyžaduje předpoklad konstantní rychlosti - to znamená, že předpokládá ultrametrický strom, ve kterém jsou vzdálenosti od kořene ke každému konci větve stejné. Když hroty jsou molekulární data (tj., DNA, RNA a protein ), které byly odebrány současně, ultrametricita předpoklad se stává ekvivalentem předpokladu a molekulární hodiny.
Pracovní příklad
Tento pracovní příklad je založen na a JC69 genetická matice vzdálenosti počítaná z 5S ribozomální RNA seřazení sekvencí pěti bakterií: Bacillus subtilis (), Bacillus stearothermophilus (), Lactobacillus viridescens (), Acholeplasma modicum (), a Micrococcus luteus ().[3][4]





První krok
- První shlukování
Předpokládejme, že máme pět prvků a následující matice párových vzdáleností mezi nimi:


| A | b | C | d | E | |
|---|---|---|---|---|---|
| A | 0 | 17 | 21 | 31 | 23 |
| b | 17 | 0 | 30 | 34 | 21 |
| C | 21 | 30 | 0 | 28 | 39 |
| d | 31 | 34 | 28 | 0 | 43 |
| E | 23 | 21 | 39 | 43 | 0 |
V tomto příkladu je nejmenší hodnota , takže spojujeme prvky a .

- Odhad délky první větve
Nechat označte uzel, ke kterému a jsou nyní připojeni. Nastavení zajišťuje, že prvky a jsou ve stejné vzdálenosti od . To odpovídá očekávání ultrametricita hypotéza. Spojení větví a na pak mají délky (viz závěrečný dendrogram )



- První aktualizace matice vzdálenosti
Poté pokračujeme v aktualizaci počáteční matice vzdálenosti do nové matice vzdálenosti (viz níže), zmenšená o jeden řádek a jeden sloupec kvůli shlukování s Tučné hodnoty v odpovídají novým vzdálenostem vypočítaným podle průměrné vzdálenosti mezi každým prvkem prvního klastru a každý ze zbývajících prvků:





Kurzíva hodnoty v nejsou ovlivněny aktualizací matice, protože odpovídají vzdálenostem mezi prvky, které nejsou zahrnuty v prvním klastru.
Druhý krok
- Druhé shlukování
Nyní zopakujeme tři předchozí kroky, počínaje novou maticí vzdálenosti
| (a, b) | C | d | E | |
|---|---|---|---|---|
| (a, b) | 0 | 25.5 | 32.5 | 22 |
| C | 25.5 | 0 | 28 | 39 |
| d | 32.5 | 28 | 0 | 43 |
| E | 22 | 39 | 43 | 0 |
Tady, je nejmenší hodnota , takže se připojíme ke klastru a prvek .

- Odhad délky druhé větve
Nechat označte uzel, ke kterému a jsou nyní připojeni. Z důvodu omezení ultrametricity se větve spojují nebo na , a na jsou stejné a mají následující délku:


Dedukujeme délku chybějící větve: (viz závěrečný dendrogram )

- Aktualizace druhé matice vzdálenosti
Poté pokračujeme v aktualizaci do nové matice vzdálenosti (viz níže), zmenšená o jeden řádek a jeden sloupec kvůli shlukování s . Tučné hodnoty v odpovídají novým vzdálenostem vypočítaným podle proporcionální průměrování:


Díky tomuto proporcionálnímu průměru odpovídá výpočet této nové vzdálenosti větší velikosti shluk (dva prvky) s ohledem na (jeden prvek). Podobně:

Proporcionální průměrování proto dává stejnou váhu počátečním vzdálenostem matice . To je důvod, proč metoda je nevážený, ne s ohledem na matematický postup, ale s ohledem na počáteční vzdálenosti.
Třetí krok
- Třetí shlukování
Znovu opakujeme tři předchozí kroky, počínaje aktualizovanou maticí vzdálenosti .
| ((a, b), e) | C | d | |
|---|---|---|---|
| ((a, b), e) | 0 | 30 | 36 |
| C | 30 | 0 | 28 |
| d | 36 | 28 | 0 |
Tady, je nejmenší hodnota , takže spojujeme prvky a .

- Odhad délky třetí větve
Nechat označte uzel, ke kterému a jsou nyní připojeny a na pak mají délky (viz závěrečný dendrogram )


- Aktualizace třetí matice vzdálenosti
Je třeba aktualizovat jeden záznam, který má na paměti tyto dva prvky a každý má příspěvek ve výši v průměrný výpočet:


Poslední krok
Finále matice je:

| ((a, b), e) | (CD) | |
|---|---|---|
| ((a, b), e) | 0 | 33 |
| (CD) | 33 | 0 |
Takže se připojujeme ke klastrům a .


Nechat označit (kořenový) uzel, ke kterému a jsou nyní připojeny a na pak mají délky:


Dedukujeme dvě zbývající délky větví:


Dendrogram UPGMA
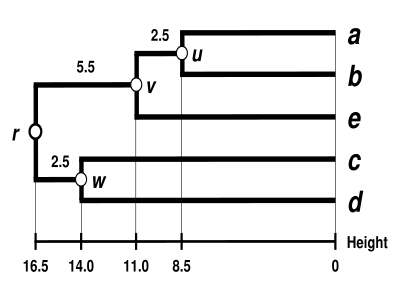
Dendrogram je nyní dokončen.[5] Je to ultrametrické, protože všechny tipy ( na ) jsou ve stejné vzdálenosti od :

Dendrogram je tedy zakořeněn pomocí , jeho nejhlubší uzel.
Srovnání s jinými vazbami
Mezi alternativní schémata propojení patří shlukování s jediným spojením, kompletní shlukování vazeb, a Průměrné shlukování propojení WPGMA. Implementace jiného propojení je jednoduše otázkou použití jiného vzorce pro výpočet meziklastrových vzdáleností během kroků aktualizace matice vzdálenosti výše uvedeného algoritmu. Kompletní shlukování vazeb se vyhne nevýhodě alternativní metody shlukování jednoho propojení - tzv fenomén řetězení, kde mohou být klastry vytvořené prostřednictvím seskupení s jednou vazbou vynuceny společně kvůli tomu, že jednotlivé prvky jsou blízko u sebe, i když mnoho prvků v každém klastru může být od sebe velmi vzdálených. Kompletní propojení má tendenci najít kompaktní shluky přibližně stejných průměrů.[6]
 |  |  |  |
| Seskupování pomocí jednoho propojení. | Kompletní shlukování. | Průměrné shlukování vazeb: WPGMA. | Průměrné shlukování vazeb: UPGMA. |
Použití
- v ekologie, je to jedna z nejpopulárnějších metod pro klasifikaci jednotek vzorkování (například vegetačních ploch) na základě jejich párových podobností v příslušných proměnných deskriptorů (jako je druhové složení).[7] Například se používá k pochopení trofické interakce mezi mořskými bakteriemi a protisty.[8]
- v bioinformatika, UPGMA se používá pro vytvoření fenetický stromy (fenogramy). UPGMA byl původně navržen pro použití v elektroforéza bílkovin studií, ale v současné době se nejčastěji používá k výrobě vodicích stromů pro sofistikovanější algoritmy. Tento algoritmus se používá například v zarovnání sekvence postupy, protože navrhuje jedno pořadí, ve kterém budou sekvence zarovnány. Cílem průvodce je seskupit nejpodobnější sekvence bez ohledu na jejich evoluční rychlost nebo fylogenetické spříznění, a to je přesně cíl UPGMA.[9]
- v fylogenetika, UPGMA předpokládá konstantní rychlost vývoje (hypotéza molekulárních hodin ) a že všechny sekvence byly vzorkovány současně a nejde o dobře pokládanou metodu pro odvození vztahů, pokud tento předpoklad nebyl testován a odůvodněn pro použitý soubor dat. Všimněte si, že ani za „přísných hodin“ by sekvence vzorkované v různých časech neměly vést k ultrametrickému stromu.
Časová složitost
Triviální implementace algoritmu pro konstrukci stromu UPGMA má časová složitost a použití haldy pro každý klastr k udržení jeho vzdáleností od jiného klastru snižuje jeho čas na . Fionn Murtagh představil některé další přístupy pro speciální případy, a časový algoritmus Day a Edelsbrunner[10] pro k-rozměrná data, která jsou optimální pro konstantu k a další algoritmus pro omezené vstupy, když „aglomerační strategie splňuje vlastnost redukovatelnosti“.[11]




Viz také
- Soused se připojil
- Shluková analýza
- Seskupování pomocí jednoho propojení
- Kompletní shlukování
- Hierarchické shlukování
- Modely evoluce DNA
- Molekulární hodiny
Reference
- ^ Sokal, Michener (1958). „Statistická metoda pro hodnocení systematických vztahů“. Vědecký bulletin University of Kansas. 38: 1409–1438.
- ^ Garcia S, Puigbò P. "DendroUPGMA: Dendrogramový stavební nástroj" (PDF). p. 4.
- ^ Erdmann VA, Wolters J (1986). "Sbírka publikovaných 5S, 5,8S a 4,5S ribosomálních RNA sekvencí". Výzkum nukleových kyselin. 14 Suppl (Suppl): r1–59. doi:10.1093 / nar / 14.suppl.r1. PMC 341310. PMID 2422630.
- ^ Olsen GJ (1988). "Fylogenetická analýza pomocí ribozomální RNA". Metody v enzymologii. 164: 793–812. doi:10.1016 / s0076-6879 (88) 64084-5. PMID 3241556.
- ^ Swofford DL, Olsen GJ, Waddell PJ, Hillis DM (1996). "Fylogenetická inference". V Hillis DM, Moritz C, Mable BK (eds.). Molekulární systematika, 2. vydání. Sunderland, MA: Sinauer. 407–514. ISBN 9780878932825.
- ^ Everitt, B. S .; Landau, S .; Leese, M. (2001). Shluková analýza. 4. vydání. Londýn: Arnold. p. 62–64.
- ^ Legendre P, Legendre L (1998). Numerická ekologie. Vývoj environmentálního modelování. 20 (Second English ed.). Amsterdam: Elsevier.
- ^ Vázquez-Domínguez E, Casamayor EO, Català P, Lebaron P (duben 2005). „Různé mořské heterotrofní nanoflageláty ovlivňují rozdílně složení obohacených bakteriálních komunit“. Mikrobiální ekologie. 49 (3): 474–85. doi:10.1007 / s00248-004-0035-5. JSTOR 25153200. PMID 16003474. S2CID 22300174.
- ^ Wheeler TJ, Kececioglu JD (červenec 2007). „Vícenásobné zarovnání zarovnáním zarovnání“. Bioinformatika. 23 (13): i559–68. doi:10.1093 / bioinformatika / btm226. PMID 17646343.
- ^ Den WH, Edelsbrunner H (01.12.1984). "Efektivní algoritmy pro aglomerativní hierarchické metody shlukování". Journal of Classification. 1 (1): 7–24. doi:10.1007 / BF01890115. ISSN 0176-4268. S2CID 121201396.
- ^ Murtagh F (1984). "Složitost hierarchických shlukovacích algoritmů: stav techniky". Výpočetní statistiky čtvrtletně. 1: 101–113.
externí odkazy
- Implementace klastrového algoritmu UPGMA v Ruby (AI4R)
- Příklad výpočtu UPGMA pomocí matice podobnosti
- Příklad výpočtu UPGMA pomocí matice vzdálenosti
| Relevantní pole | ||
|---|---|---|
| Základní pojmy | ||
| Inferenční metody | ||
| Aktuální témata | ||
| Skupinové vlastnosti | ||
| Skupinové typy | ||
| Nomenklatura | ||
| ||