MPI-CDG - MPI-CDG
| MPI-CDG | |
|---|---|
| Ostatní jména | CDG-IB |
 | |
| Specialita | Lékařská genetika |
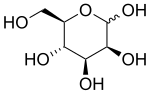
| Léčba | doplnění manózy |
MPI-CDG je autozomálně recesivní vrozená porucha glykosylace způsobené bialelickými patogenními variantami v MPI. Klinické příznaky v MPI-CDG jsou způsobeny nedostatečnou aktivitou enzymu manóza fosfát izomeráza. Klinicky jsou nejčastější příznaky MPI-CDG chronické průjem, neprospívání, enteropatie ztráta bílkovin a koagulopatie.[1] MPI-CDG se liší od většiny ostatních popsaných poruch glykosylace kvůli tomu, že chybí zapojení centrálního nervového systému a protože má kromě podpůrné péče i možnosti léčby. Doplnění orálně manóza Bylo prokázáno, že zlepšuje většinu příznaků onemocnění. Pokud není léčen, může být MPI-CDG smrtelný.[1] MPI-CDG byl dříve známý jako CDG-IB. Porucha byla poprvé klinicky popsána v roce 1986 a základní genetická vada byla identifikována v roce 1998.[1][2]
Reference
- ^ A b C „# 602579 KONGENITÁLNÍ PORUCHA GLYKOSYLACE, TYP Ib; CDG1B“. Univerzita Johna Hopkinse. Citováno 2019-04-30.
- ^ „MPI-CDG (CDG-Ib) | Informační centrum o genetických a vzácných onemocněních (GARD) - program NCATS“. rarediseases.info.nih.gov. Citováno 1. května 2019.