Katalýza fotoredoxem - Photoredox catalysis
Katalýza fotoredoxem je pobočkou katalýza který využívá energii světlo zrychlit a chemická reakce přes přenos jednoho elektronu Události.[1][2][3][4][5] Tato oblast je pojmenována jako kombinace „fotografie“ odkazující na světlo a redox, zhuštěný výraz pro chemické procesy snížení a oxidace. Zejména fotoredoxní katalýza využívá malá množství sloučeniny citlivé na světlo, která při excitaci světlem může zprostředkovat přenos elektrony mezi chemickými sloučeninami, které by obvykle vůbec nereagovaly. Katalyzátory fotoredoxu jsou obecně čerpány ze tří tříd materiálů: komplexy přechodných kovů, organická barviva a polovodiče. Zatímco v devadesátých a na počátku dvacátých let dominovaly organické katalyzátory fotoredoxu,[6] dnes se běžněji používají rozpustné komplexy přechodného kovu.
![Schematické znázornění [Ru (bipy) 3] 2+, typického katalyzátoru fotoredoxu](http://upload.wikimedia.org/wikipedia/commons/thumb/3/32/Ru%28bipy%29_Schematic.png/220px-Ru%28bipy%29_Schematic.png)
Studium tohoto odvětví katalýzy vedlo k vývoji nových metod k dosažení známých a nových chemických transformací. Katalyzátory fotoredoxu jsou obvykle mnohem méně toxické než tradiční činidla používaná ke generování volné radikály, jako organický cín sloučeniny. Kromě toho fotoredoxní katalyzátory vytvářejí při působení světla silná redoxní činidla, jsou za normálních podmínek nereaktivní. Katalyzátory fotoredoxu s komplexem přechodného kovu jsou tedy atraktivnější než stechiometrický redoxní látky jako chinony. Vlastnosti katalyzátorů fotoredoxu s přechodovým kovem závisí na ligandech a kovu a lze je modifikovat pro různé účely.
Photoredoxová katalýza se často používá k vytváření známých reaktivních meziproduktů novým způsobem a vedla k objevu nových organických reakcí, jako je například první přímá funkcionalizace β-arylace nasycených aldehydy. Zatímco D3-symetrické komplexy přechodných kovů používané v mnoha reakcích katalyzovaných fotoredoxem jsou chirální, enantioenrichtované fotoredoxní katalyzátory vedly pouze k nízkým hladinám enantioselektivita při fotoredoxované katalyzované aryl-arylové kopulační reakci, což naznačuje, že chirální povaha těchto katalyzátorů je stále špatná při přenosu stereochemický informace.[7] I když syntetické užitečné úrovně enantioselektivity nebyly dosaženy pouze za použití chirálních katalyzátorů fotoredoxu, enantioselektivita byla získána synergickou kombinací fotoredox katalýzy s chirálními organokatalyzátory, jako jsou sekundární aminy a Bronstedovy kyseliny.[8]
Fotochemie senzitizátorů přechodových kovů
Senzibilizátory absorbují světlo a vytvářejí redoxně aktivní excitované stavy. U mnoha senzibilizátorů na bázi kovu je excitace realizována jako a přenos náboje kov na ligand, přičemž elektron se pohybuje z kovu (např. d orbital) na orbitál lokalizovaný na ligandech (např. π * orbitální aromatického ligandu). Počáteční excitovaný elektronický stav se uvolní do excitovaného stavu singletu s nejnižší energií interní konverze, proces, při kterém je energie rozptýlena spíše jako vibrační energie než jako elektromagnetické záření. Tento vzrušený stav singletu se může dále uvolnit dvěma odlišnými procesy: katalyzátor může fluoreskují, vyzařující foton a vracející se do základního stavu singletu, nebo se může přesunout do stavu excitace tripletů s nejnižší energií (stav, kdy mají dva nepárové elektrony stejnou rotaci) druhým neradiačním procesem nazývaným křížení mezi systémy.
Přímá relaxace excitovaného tripletu do základního stavu, tzv fosforescence, vyžaduje jak emisi fotonu, tak inverzi rotace excitovaného elektronu. Tato cesta je pomalá, protože je zakázáno otáčení takže vzrušený stav tripletů má podstatnou průměrnou životnost. U běžného fotosenzibilizátoru tris- (2,2’-bipyridyl) ruthenium (ve zkratce [Ru (bipy)3]2+ nebo [Ru (bpy)3]2+), životnost excitovaného stavu tripletů je přibližně 1100 ns. Tato životnost je dostatečná pro to, aby se před rozpadem katalyzátoru do základního stavu vyskytly další relaxační dráhy (konkrétně dráhy přenosu elektronů).

Dlouhodobý vzrušený stav tripletů přístupný fotoexcitací je účinnější redukční činidlo a silnější oxidační činidlo než základní stav katalyzátoru. Protože senzibilizátor je koordinačně nasycen, musí k přenosu elektronu dojít pomocí vnější koule proces, kde elektron tunely mezi katalyzátorem a substrátem.
Přenos elektronů vnější sféry
Marcus ' teorie přenosu elektronů vnější sféry předpovídá, že takový tunelovací proces nastane nejrychleji v systémech, kde je přenos elektronů termodynamicky příznivý (tj. mezi silnými redukčními činidly a oxidanty) a kde má přenos elektronů nízkou vnitřní bariéru.
Vnitřní bariéra přenosu elektronů pochází z Princip Franck – Condon, s uvedením, že elektronický přechod probíhá rychleji vzhledem k většímu překrývání mezi počátečním a konečným elektronickým stavem. Tento princip, interpretovaný volně, naznačuje, že bariéra elektronického přechodu souvisí s mírou, do jaké se systém snaží reorganizovat. U elektronového přechodu se systémem bariéra souvisí s „překrytím“ mezi počáteční a konečnou vlnovou funkcí excitovaného elektronu - tj. míra, do jaké se elektron musí při přechodu „pohybovat“.
V mezimolekulárním přenosu elektronů hraje podobnou roli míra, do jaké se jádra snaží pohybovat v reakci na změnu v jejich novém elektronickém prostředí. Ihned po přenosu elektronů představuje jaderné uspořádání molekuly, dříve rovnovážné, nyní vibračně vzrušený stav a musí se uvolnit ve své nové rovnovážné geometrii. Pevné systémy, jejichž geometrie není do značné míry závislá na oxidačním stavu, proto během přenosu elektronů zažívají menší vibrační buzení a mají nižší vnitřní bariéru. Fotokatalyzátory jako [Ru (bipy)3]2+, jsou drženy v tuhém uspořádání plochými bidentátními ligandy uspořádanými v osmistěn geometrie kolem kovového středu. Komplex proto během přenosu elektronů moc neorganizuje. Protože elektronový přenos těchto komplexů je rychlý, je pravděpodobné, že k němu dojde během trvání aktivního stavu katalyzátoru, tj. Během života excitovaného stavu tripletů.

Regenerace katalyzátoru
Posledním krokem ve fotokatalytickém cyklu je regenerace fotokatalyzátoru v jeho základním stavu. V této fázi katalyzátor existuje jako základní stav svých oxidovaných nebo redukovaných forem, v závislosti na tom, zda daroval nebo přijal elektron. Tyto oxidační stavy mají silnou hnací sílu k návratu do rovnovážného oxidačního stavu a působí jako silné jednoelektronové redukční činidlo nebo oxidační činidlo k uspokojení této hnací síly.
K regeneraci původního základního stavu se katalyzátor musí účastnit druhého přenosu elektronů vnější koulí. V mnoha případech se tento přenos elektronů provádí stechiometrickým dvouelektronovým redukčním činidlem nebo oxidantem, i když v některých případech tento krok zahrnuje druhé činidlo. Redukční kalicí cyklus je, když je katalyzátor excitovaného stavu nejprve redukován a poté oxidován, aby se vrátil do klidového stavu. Naopak, oxidační zhášecí cyklus je, když je katalyzátor excitovaného stavu nejprve oxidován a poté redukován, aby se vrátil do klidového stavu. Tyto dva cykly lze odlišit a Stern – Volmerův experiment.
Protože krok přenosu elektronů v katalytickém cyklu probíhá z excitovaného stavu tripletů, soutěží s fosforescencí jako relaxační cestou. Stern – Volmerův experiment měří intenzitu fosforescence a mění koncentraci každého možného zhášecího činidla. Když se mění koncentrace skutečného zhášecího činidla, je ovlivněna rychlost přenosu elektronů a stupeň fosforescence. Tento vztah je modelován rovnicí:
![left ({ frac {I_ {0}} {I}} right) = 1 + {k_ {q}} * { tau _ {0}} times [Q]](https://wikimedia.org/api/rest_v1/media/math/render/svg/338eb04d84052783d691791ccf5c329070594aa0)
Tady, já0 a označuji intenzitu emise s přítomným nebo bez přítomného zhášecího činidla, kq rychlostní konstanta procesu kalení, τ0 životnost excitovaného stavu v nepřítomnosti zhášecího činidla a [Q] koncentrace zhášecího činidla. Pokud je tedy životnost excitovaného stavu katalyzátoru fotoredoxu známa z jiných experimentů, lze rychlostní konstantu zhášení za přítomnosti jedné reakční složky určit měřením změny intenzity emisí při změně koncentrace zhášecího činidla.
Fotofyzikální vlastnosti
Redoxní potenciály
Redoxní potenciály fotoredoxních katalyzátorů musí být přizpůsobeny dalším složkám reakce. Zatímco redoxní potenciály základního stavu lze snadno měřit pomocí cyklická voltametrie nebo jinými elektrochemickými metodami, měření redoxního potenciálu elektronicky excitovaného stavu nelze těmito metodami dosáhnout přímo.[9] Existují však dvě metody, které umožňují odhad redoxních potenciálů v excitovaném stavu, a existuje jedna metoda pro přímé měření těchto potenciálů. Pro odhad redoxních potenciálů v excitovaném stavu je jednou metodou srovnání rychlostí přenosu elektronů z excitovaného stavu do řady reaktantů v základním stavu, jejichž redox potenciály jsou známy. Běžnější metodou pro odhad těchto potenciálů je použití rovnice vyvinuté Rehmem a Wellerem, která popisuje potenciály excitovaného stavu jako korekci potenciálů základního stavu:


V těchto vzorcích je E *1/2 představuje redukční nebo oxidační potenciál excitovaného stavu, E.1/2 představuje redukční nebo oxidační potenciál základního stavu, E0,0 představuje rozdíl v energii mezi nulami vibračních stavů země a excitovaných stavů a wr představuje pracovní funkce, elektrostatická interakce, která vzniká v důsledku oddělení nábojů, ke kterým dochází při přenosu elektronů mezi dvěma chemickými látkami. Nulová-nulová excitační energie, E0,0 je obvykle aproximován odpovídajícím přechodem ve fluorescenčním spektru. Tato metoda umožňuje výpočet přibližných redoxních potenciálů excitovaného stavu ze snadněji měřitelných redoxních potenciálů základního stavu a spektroskopických dat.
Přímé měření redoxních potenciálů v excitovaném stavu je možné použitím metody známé jako fázově modulované voltametrie. Tato metoda funguje tak, že svítí světlo na elektrochemický článek za účelem generování požadovaných druhů v excitovaném stavu, ale za účelem modulace intenzity světla sinusově, takže koncentrace druhu excitovaného stavu není konstantní. Ve skutečnosti by se koncentrace druhů v excitovaném stavu v buňce měla měnit přesně ve fázi s intenzitou světla dopadajícího na elektrochemický článek. Pokud je potenciál aplikovaný na buňku dostatečně silný na to, aby došlo k přenosu elektronů, lze změřit koncentraci redoxně kompetentního excitovaného stavu jako střídavý proud (AC). Kromě toho fázový posun střídavého proudu vzhledem k intenzitě dopadajícího světla odpovídá průměrné životnosti druhu excitovaného stavu před tím, než se zapojí do přenosu elektronů.
Pro rychlý přístup jsou k dispozici grafy redoxních potenciálů pro nejběžnější katalyzátory fotoredoxu.[10]
Ligandová elektronegativita
Relativní redukční a oxidační povahu těchto fotokatalyzátorů lze pochopit zvážením elektronegativity ligandů a kovového centra katalyzátorového komplexu. Více elektronegativních kovů a ligandů může stabilizovat elektrony lépe než jejich méně elektronegativní protějšky. Komplexy s více elektronegativními ligandy jsou proto více oxidující než méně elektronegativní komplexy ligandů. Například ligandy 2,2'-bipyridin a 2,2'-fenylpyridin jsou izoelektronické struktury, které obsahují stejný počet a uspořádání elektronů. Fenylpyridin nahradí jeden z atomů dusíku v bipyridinu atomem uhlíku. Uhlík je méně elektronegativní než dusík, takže drží elektrony méně pevně. Jelikož je zbytek molekuly ligandu identický a fenylpyridin drží elektrony méně pevně než bipyridin, je silněji donorný elektrony a méně elektronegativní jako ligand. Komplexy s fenylpyridinovými ligandy proto silněji redukují a méně silně oxidují než ekvivalentní komplexy s bipyridinovými ligandy.
Podobně je fluorovaný fenylpyridinový ligand elektronegativnější než fenylpyridin, takže komplexy s ligandy obsahujícími fluor jsou silněji oxidující a méně silně redukující než ekvivalentní nesubstituované fenylpyridinové komplexy. Elektronický vliv kovového centra na komplex je složitější než účinek ligandu. Podle Paulingova stupnice elektronegativity, obojí ruthenium a iridium mít elektronegativitu 2,2. Pokud by to byl jediný faktor relevantní pro redoxní potenciály, pak by komplexy ruthenia a iridia se stejnými ligandy měly být stejně silnými katalyzátory fotoredoxu. Vzhledem k Rehm-Wellerově rovnici však spektroskopické vlastnosti kovu hrají roli při určování redoxních vlastností excitovaného stavu.[11] Zejména parametr E0,0 souvisí s emisní vlnovou délkou komplexu, a proto s velikostí Stokesova posunu - rozdílu v energii mezi maximální absorpcí a emisí molekuly. Rutheniové komplexy mají obvykle velké Stokesovy posuny, a proto mají ve srovnání s iridiovými komplexy nízké vlnové délky emise energie a malé nulové excitační energie. Ve skutečnosti, zatímco komplexy ruthenia v základním stavu mohou být účinnými redukčními činidly, komplex excitovaného stavu je mnohem méně účinným redukčním činidlem nebo oxidačním činidlem než jeho ekvivalentní komplex iridia. Díky tomu je iridium výhodné pro vývoj obecných organických transformací, protože silnější redoxní potenciály excitovaného katalyzátoru umožňují použití slabších stechiometrických redukčních činidel a oxidantů nebo použití méně reaktivních substrátů.[11]
Aplikace
Redukční dehalogenace
Redukční dehalogenace je odstranění halogen atomy z molekuly. Tradiční metoda dehalogenace však používá stechiometrická organocínová činidla, jako je např tributylcín hydrid. I když je tato reakce silná s vysokou funkční skupina tolerance, organotinová činidla jsou vysoce toxická. Štěpení aktivovaných a reduktivně labilních funkčních skupin, včetně sulfonií a halogenů, je nejčasnější aplikací fotoredoxní katalýzy na organickou syntézu, ale první pokusy byly omezeny potřebou specifických substrátů nebo tvorbou dimerních kopulačních produktů.[12][13][14][15][16] Obecnější metody jsou známé.[17] Jedna metoda využívá [Ru (bipy)3]2+ jako fotokatalyzátor a stechiometrické aminové redukční činidlo ke snížení "aktivovaných" vazeb uhlík-halogen, jako jsou vazby se sousední karbonylovou skupinou nebo arenem. Tyto vazby jsou považovány za aktivované, protože radikál, který produkují při fragmentaci, je stabilizován konjugací s karbonylovou skupinou nebo arenem. Stechiometrické redukční činidlo přítomné v této reakci přenáší elektron k redukci katalyzátoru excitovaného stavu na oxidační stav Ru (I). Redukovaný katalyzátor poté převede přenesený elektron na halogenovaný substrát, čímž sníží slabou vazbu C-X a vyvolá fragmentaci.
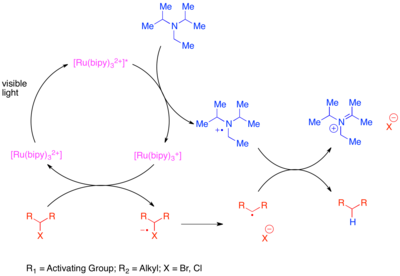
Neaktivované vazby uhlík-jod lze snížit pomocí silně redukujícího fotokatalyzátoru tris- (2,2’-fenylpyridin ) iridium (Ir (ppy)3).[18] Tato reakce je mechanicky odlišná od předchozí transformace aktivovaných bromidů a chloridů. Zvýšený redukční potenciál Ir (ppy)3 ve srovnání s [Ru (bipy)3]2+ umožňuje přímou redukci vazby uhlík-jod bez interakce se stechiometrickým redukčním činidlem. Iridiový komplex tedy přenáší elektron na substrát, což způsobuje fragmentaci substrátu a oxidaci katalyzátoru na oxidační stav Ir (IV). Oxidovaný fotokatalyzátor se vrací do původního oxidačního stavu oxidací reakčních přísad.

Stejně jako radikálovými dehalogenačními reakcemi zprostředkovanými cínem lze použít fotokatalytickou redukční dehalogenaci k zahájení kaskádové cyklizace k rychlému vytvoření molekulární složitosti.[19] V této práci došlo k radikální kaskádové cyklizaci, která uzavřela dva pětičlenné kruhy a vytvořila dvě nová stereocentra, s dobrým výtěžkem. Tento protokol reduktivní dehalogenace byl klíčovým krokem v celkové syntéze přírodního produktu (+) - Gliocladinu C.[20]

Oxidační generace iminiových iontů
Iminium ionty jsou silné elektrofily užitečné pro generování C-N vazeb ve složitých molekulách. Kondenzace z aminy s karbonyl sloučeniny za vzniku iminiových iontů je často nepříznivé, někdy vyžaduje drsné dehydratační podmínky. Cenným nástrojem syntézy jsou tedy alternativní metody pro generování iminiových iontů, zejména oxidací z odpovídajícího aminu. Iminové ionty lze generovat z aktivovaných aminů pomocí Ir (dtbbpy) (ppy)2PF6 jako katalyzátor fotoredoxu.[21] Předpokládá se, že k této transformaci dochází oxidací aminu na aminium radikální kation vzrušený fotokatalyzátor. Poté následuje přenos atomu vodíku na superstechimetrický oxidant, jako je trichlormethylový radikál (CCI3 za vzniku iminiového iontu). Iminiový iont se poté rozloží reakcí s nukleofilem. Související transformace aminů s celou řadou dalších nukleofily byly vyšetřovány, jako např kyanid (Streckerova reakce ), silyl enol ethery (Mannichova reakce ), dialkylfosfáty, allylsilany (aza-Sakurai reakce ), indoly (Friedel-Craftsova reakce ) a acetylidy mědi.[22][23][24][25][26]

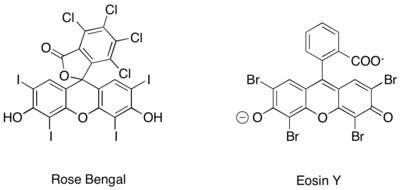
Podobná generace fotoredoxu iminiových iontů byla dále dosažena za použití čistě organických katalyzátorů fotoredoxu, jako je například Rose Bengal a Eosin Y.[27][28][29]
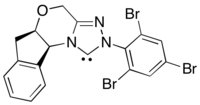
Asymetrická varianta této reakce využívá acylové nukleofilní ekvivalenty generované N-heterocyklický karben katalýza.[30] Tato reakční metoda obchází problém špatné enantioindukce z chirálních fotoredoxních katalyzátorů přesunutím zdroje enantioselektivity na N-heterocyklický karben.
Oxidační tvorba oxokarbenia iontů
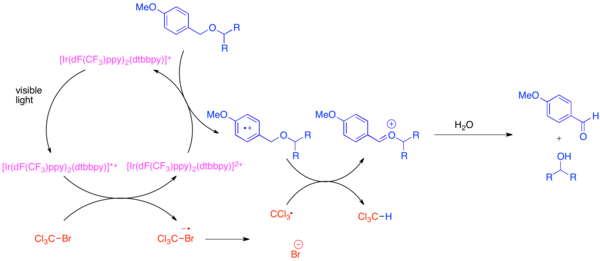
Vývoj ortogonálních chránících skupin je problémem v organické syntéze, protože tyto chránící skupiny umožňují každou instanci společné funkční skupiny, jako je hydroxyl během syntézy komplexní molekuly. Velmi běžnou chránící skupinou pro hydroxylovou funkční skupinu je odst-methoxybenzyl (PMB) ether. Tato chránící skupina je chemicky podobná benzyletheru méně bohatému na elektrony. Typické selektivní štěpení PMB etheru v přítomnosti benzyletheru používá silná stechiometrická oxidační činidla, jako je 2,3-dichlor-5,6-dikyano-l, 4-benzochinon (DDQ) nebo ceričnan amonný (UMĚT). Ethery PMB jsou mnohem náchylnější k oxidaci než benzylethery, protože jsou bohatší na elektrony. Selektivní deprotekce etherů PMB lze dosáhnout použitím bis- (2- (2 ', 4'-difluorfenyl) -5-trifluormethylpyridin) - (4,4'-ditertbutylbipyridin) iridium (III) hexafluorfosfátu (Ir [dF (CF3) ppy]2(dtbbpy) PF6) a mírné stechiometrické oxidační činidlo, jako je bromtrichlormethan, BrCCl3.[31] Fotoexcitovaný iridiový katalyzátor se dostatečně redukuje na fragmentaci bromtrichlormethanu za vzniku trichlormethylového radikálu, bromidového aniontu a komplexu Ir (IV). Elektronově chudé fluorované ligandy způsobují, že iridiový komplex dostatečně oxiduje, aby přijal elektron z arenu bohatého na elektrony, jako je ether PMB. Poté, co je aren oxidován, bude se snadno účastnit přenosu atomu vodíku trichlormethylovým radikálem za vzniku chloroformu a oxokarbenium iont, který se snadno hydrolyzuje, aby se uvolnil volný hydroxid. Ukázalo se, že tato reakce je ortogonální k mnoha běžným ochranným skupinám, když byla přidána báze k neutralizaci produkovaného HBr.
Cykloadice
Cykloadice a další pericyklické reakce jsou silné transformace v organické syntéze kvůli jejich potenciálu rychle generovat složité molekulární architektury a zejména kvůli jejich schopnosti nastavit více sousedících stereocentra vysoce kontrolovaným způsobem. Podle teplotních podmínek jsou však povoleny pouze určité cykloadice Woodward – Hoffmann vládne orbitální symetrie nebo jiné ekvivalentní modely jako např hraniční molekulární orbitální teorie (FMO) nebo model Dewar-Zimmermann. Cykloadice, které nejsou tepelně povoleny, jako je cykloadice [2 + 2], lze aktivovat fotochemickou aktivací reakce. Za nekatalyzovaných podmínek vyžaduje tato aktivace použití vysoké energie ultrafialové světlo schopné změnit orbitální populace reaktivních sloučenin. Alternativně se uvádí, že kovové katalyzátory, jako je kobalt a měď, katalyzují tepelně zakázané [2 + 2] cykloadice pomocí přenosu jediného elektronu.

Požadované změny v orbitálních populacích lze dosáhnout přenosem elektronů s fotokatalyzátorem citlivým na viditelné světlo s nižší energií.[32][33][34][35][36] Yoon prokázal účinné intra- a intermolekulární [2 + 2] cykloadice aktivovaných olefiny: zvláště enones a styreny. Bylo zjištěno, že enony nebo olefiny chudé na elektrony reagují cestou radikálově-aniontů s využitím diisopropylethylamin jako přechodný zdroj elektronů. Pro tento elektronový přenos [Ru (bipy)3]2+ byl objeven jako účinný fotokatalyzátor. Aniontová povaha cyklizace se ukázala jako zásadní: provedení reakce v kyselině spíše než s lithiovým protiiionem upřednostňovalo necykloadiční dráhu.[37] Zhao a kol. rovněž zjistil, že je k dispozici stále odlišná cyklická cesta chalcones s samarium protiion.[38] Naopak bylo zjištěno, že styreny bohaté na elektrony reagují pomocí mechanismu radikálových kationů s využitím methyl viologen nebo molekulární kyslík jako přechodný elektronový jímka. While [Ru (bipy)3]2+ se ukázalo být kompetentním katalyzátorem pro použití intramolekulárních cyklizací methyl viologen, nemohl být použit s molekulárním kyslíkem jako jímkou elektronů nebo pro intermolekulární cyklizace. Pro intermolekulární cyklizaci Yoon et al. objevil, že silněji oxidující fotokatalyzátor [Ru (bpm)3]2+ a molekulární kyslík poskytly katalytický systém, který je vhodnější pro přístup k radikálnímu kationtu nezbytnému pro cykloadici. [Ru (bpz)3]2+, stále silněji oxidující fotokatalyzátor, se ukázal jako problematický, protože i když mohl katalyzovat požadovanou [2 + 2] cykloadici, byl také dostatečně silný, aby oxidoval cykloadid a katalyzoval retro- [2 + 2] reakci. Toto srovnání fotokatalyzátorů zdůrazňuje důležitost vyladění redoxních vlastností fotokatalyzátoru na reakční systém a také prokázání hodnoty polypyridylových sloučenin jako ligandů vzhledem k tomu, jak snadno je lze modifikovat, aby se upravily redoxní vlastnosti jejich komplexů.

Cykloadice katalyzované fotoredoxem [2 + 2] mohou být také provedeny s trifenylpyrylovým organickým fotoredoxním katalyzátorem.[39]

Kromě tepelně zakázané cykloadice [2 + 2] lze na cyklizaci [4 + 2] použít fotoredoxní katalýzu (Diels-Alderova reakce ). Bis-enony, podobné substrátům použitým pro cyklizaci fotoredoxem [2 + 2], ale s delší linkerovou vazbou spojující dvě enonové funkční skupiny, podléhají intramolekulární radikál-anionové hetero-Diels-Alderovy reakce rychleji než [2 + 2] cykloadice.[40]

Podobně se styreny bohaté na elektrony účastní intra- nebo intermolekulárních cyklů Diels – Alder prostřednictvím mechanismu radikálních kationtů.[41][42] [Ru (bipy)3]2+ byl kompetentním katalyzátorem pro intermolekulární, ale ne intramolekulární Diels – Alderovy cyklizace. Tato fotoredoxem katalyzovaná Diels-Alderova reakce umožňuje cykloadici mezi dvěma elektronicky neodpovídajícími substráty. Normální elektronická poptávka po reakci Diels – Alder vyžaduje elektrony bohaté dien reagovat s elektronově chudým olefinem (nebo „dienofilem“), zatímco inverzní elektronová poptávka Diels – Alderova reakce probíhá mezi opačným případem elektronově chudého dienu a velmi elektronově bohatého dienofilu. Případ photoredox, protože probíhá jiným mechanismem než tepelná Diels-Alderova reakce, umožňuje cykloadici mezi dienem bohatým na elektrony a dienofilem bohatým na elektrony, což umožňuje přístup k novým třídám Diels-Alderových aduktů.

Syntetická hodnota Yoonovy fotoredoxem katalyzované styrenové reakce Diels – Alder byla prokázána celkovou syntézou přírodního produktu Heitziamide A.[41] Tato syntéza ukazuje, že tepelná Diels-Alderova reakce upřednostňuje nežádoucí regioisomer, ale reakce katalyzovaná fotoredoxem poskytuje požadovaný regioisomer ve zlepšeném výtěžku.

Organokatalýza Photoredox
Organokatalýza je podpole katalýzy, která zkoumá potenciál organických malých molekul jako katalyzátorů, zejména pro enantioselektivní tvorbu chirálních molekul. Jednou strategií v tomto podpole je použití chirálních sekundárních aminů k aktivaci karbonylových sloučenin. V tomto případě kondenzace aminu s karbonylovou sloučeninou generuje nukleofilní látku enamin. Chirální amin je navržen tak, aby jedna strana enaminu byla stéricky chráněna a aby pouze nestíněná strana mohla volně reagovat. Navzdory síle tohoto přístupu katalyzovat enantioselektivní funkcionalizaci karbonylových sloučenin, některé cenné transformace, jako je katalytická enantioselektivní α-alkylace aldehydy, zůstal nepolapitelný. Kombinace metod organokatalýzy a fotoredoxu poskytuje katalytické řešení tohoto problému.[43] V tomto přístupu pro a-alkylaci aldehydů, [Ru (bipy)3]2+ reduktivně fragmentuje aktivovaný alkylhalogenid, jako je bromomalonát nebo fenacylbromid, které pak mohou enantioselektivním způsobem přidat ke katalyticky generovanému enaminu. Oxidovaný fotokatalyzátor poté oxidativně uhasí výsledný a-amino radikál za vzniku iminiového iontu, který hydrolyzuje za vzniku funkcionalizované karbonylové sloučeniny. Ukázalo se, že tato transformace fotoredoxu je mechanicky odlišná od jiného organokatalytického radikálového procesu, který se nazývá singly obsazená molekulární orbitální (SOMO) katalýza. SOMO katalýza využívá superstechiometrii ceričnan amonný (CAN) oxidovat katalyticky generovaný enamin na odpovídající radikální kation, který se pak může přidat do vhodného vazebného partnera, jako je allylsilan. Tento typ mechanismu je pro fotokatalytickou alkylační reakci vyloučen, protože zatímco bylo pozorováno, že enaminový radikální kationt cykluje na závěsné olefiny a otevřené cyklopropanové radikálové hodiny v SOMO katalýze, tyto struktury byly ve fotoredoxní reakci nereaktivní.
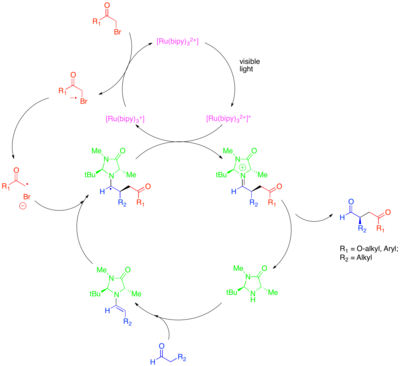
Tato transformace zahrnuje alkylace s jinými třídami aktivovaných alkylhalogenidy syntetického zájmu. Zejména použití fotokatalyzátoru Ir (dtbbpy) (ppy)2+ umožňuje enantioselektivní α-trifluormethylaci aldehydů, zatímco použití Ir (ppy)3 umožnilo enantioselektivní kopulaci aldehydů s elektronově chudými benzylbromidy.[44][45] Zeitler et al. také zkoumal produktivní sloučení fotoredoxových a organokatalytických metod k dosažení enantioselektivní alkylace aldehydů.[46] Stejný chirální imidazolidinonový organokatalyzátor byl použit k vytvoření enaminu a zavedení chirality. Místo komplexu ruthenia nebo iridia byl však použit organický katalyzátor fotoredoxu Eosin Y.
Přímá β-arylace nasycených aldehydů a ketony lze dosáhnout kombinací fotoredoxu a organokatalytických metod.[47] Předchozí metoda k dosažení přímé β-funkcionalizace nasyceného karbonylu spočívá v jednom hrnci, který se skládá ze dvoustupňového procesu, který je katalyzován sekundárním aminovým organokatalyzátorem: stechiometrická redukce aldehydu pomocí IBX s následným přidáním aktivovaného alkyl nukleofilu k beta pozici výsledného enal.[48] Tato transformace, která stejně jako jiné procesy fotoredoxu probíhá radikálním mechanismem, je omezena na přidání vysoce elektrofilních arenů do polohy beta. Závažná omezení rozsahu arenové složky v této reakci je způsobena primárně potřebou aniontu radikálu arenu, který je dostatečně stabilní, aby nereagoval přímo s enaminem nebo kationem enaminového radikálu. V navrhovaném mechanismu je aktivovaný katalyzátor fotoredoxu oxidačně uhasen arenem s nedostatkem elektronů, jako je 1,4-dikyanobenzen. Fotokatalyzátor poté oxiduje enaminové druhy, přechodně generované kondenzací aldehydu se sekundárním aminovým kokatalyzátorem, jako je optimální isopropylbenzylamin. Výsledný enaminový radikální kation obvykle reaguje jako 3 π-elektronový systém, ale vzhledem ke stabilitě radikálových vazebných partnerů vede deprotonace p-methylenové polohy k 5 π-elektronovému systému se silným radikálovým charakterem u nově přístupného β-uhlík. Ačkoli se tato reakce spoléhá na použití sekundárního aminového organokatalyzátoru pro generování enaminových druhů, které se v navrhovaném mechanismu oxidují, neexistuje žádná enantioselektivní varianta této reakce.

Vývoj této přímé β-arylace aldehydů vedl k souvisejícím reakcím na β-funkcionalizaci cyklických ketonů. Zejména β-arylace cyklických ketonů byla dosažena za podobných reakčních podmínek, ale za použití azepan jako sekundární aminový kokatalyzátor. Fotokatalytická „homo-aldolová“ reakce funguje u cyklických ketonů, což umožňuje navázání beta-polohy ketonu na ipso uhlík arylketonů, jako je benzofenon a acetofenon.[49] Kromě azepanového kokatalyzátoru vyžaduje tato reakce použití silněji redukujícího katalyzátoru fotoredoxu Ir (ppy)3 a přidání lithium hexafluoroarsenidu (LiAsF6) k podpoře redukce arylketonu jednou elektronem.
Přídavky k olefinům
Použití katalýzy fotoredoxem ke generování reaktivních radikálů zaměřených na heteroatom bylo poprvé prozkoumáno v 90. letech.[50] [Ru (bipy)3]2+ Bylo zjištěno, že katalyzuje fragmentaci tosylfenylselenidu na fenylselenolátový aniont a tosylový radikál a že mechanismus šíření radikálního řetězce umožnil přidání tosylového radikálu a fenylseleno-radikálu přes dvojnou vazbu elektronově bohatých alkylvinyletherů. Vzhledem k tomu, že fenylselenolátový anion je snadno oxidován na difenyldiselenid, byla pozorovaná nízká množství difenyldiselenidu brána jako známka toho, že fotoredoxem katalyzovaná fragmentace tosylfenylselenidu byla důležitá pouze jako iniciační krok a že většina reaktivity byla způsobena radikálním řetězovým procesem.
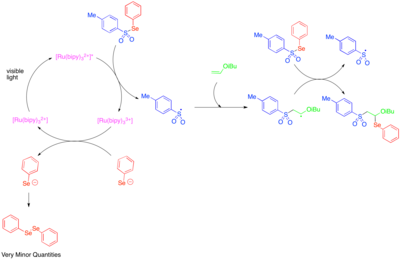
Heteroaromatické přísady k olefinům zahrnují vícesložkové oxy- a aminotrifluormethylační reakce.[51][52] Tyto reakce používají Umemotovo činidlo, sulfoniovou sůl, která slouží jako elektrofilní zdroj trifluormethylové skupiny a která předchází reakci cestou přenosu jednoho elektronu. Redukce jediného elektronu Umemotova činidla tedy uvolňuje trifluormethyl radikál, který se přidává k reaktivnímu olefinu. Následně oxidace jednoho elektronu alkylového radikálu generovaného tímto přídavkem produkuje kation, který může být zachycen vodou, alkoholem nebo nitrilem. Aby se dosáhlo vysokých úrovní regioselektivity, byla tato reaktivita zkoumána hlavně pro styreny, které jsou předpjaté směrem k tvorbě meziproduktu benzylového radikálu.

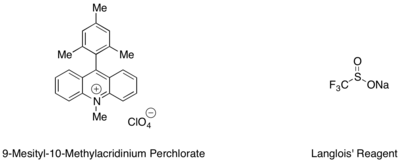
Hydrotrifluormethylace styrenů a alifatických alkenů může být provedena za použití organického fotoredoxoxidového katalyzátoru mesityl akridinium a Langloisova činidla jako zdroje CF3 radikální.[53] Při této reakci bylo zjištěno, že trifluorethanol a podstechiometrická množství aromatického thiolu, jako je methylthiosalicylát, použitého v tandemu, sloužily jako nejlepší zdroj vodíkového radikálu k dokončení katalytického cyklu.
Intramolekulární hydroetherifikace a hydroaminace probíhají s anti-Markovnikovovou selektivitou.[54][55] Jeden mechanismus vyvolává oxidaci jednoho elektronu olefinu, zachycuje kation radikálu pomocí přívěskové hydroxylové nebo aminové funkční skupiny a uhasí výsledný alkylový radikál přenosem atomu H z vysoce labilního donorového druhu. Rozšíření této reaktivity na intermolekulární systémy vedly k i) nové syntetické cestě ke komplexním tetrahydrofuranům „cyklickou cyklickou adicí polárních radikálů“ (reakce PRCC) alylalkoholu s olefinem a ii) přidáním anti-Markovnikova karboxylové kyseliny na olefiny.[56][57]
Reference
- ^ Tucker, Joseph W .; Stephenson, Corey R. J. (17. února 2012). „Zářící světlo na katalýzu fotoredoxu: teorie a syntetické aplikace“. The Journal of Organic Chemistry. 77 (4): 1617–1622. doi:10.1021 / jo202538x. PMID 22283525.
- ^ Prier, Christopher K.; Rankic, Danica A.; MacMillan, David W. C. (10 July 2013). "Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis". Chemické recenze. 113 (7): 5322–5363. doi:10.1021/cr300503r. PMC 4028850. PMID 23509883.
- ^ Yoon, Tehshik P.; Ischay, Michael A .; Du, Juana (23 June 2010). "Visible light photocatalysis as a greener approach to photochemical synthesis". Přírodní chemie. 2 (7): 527–532. doi:10.1038/NCHEM.687. PMID 20571569.
- ^ Xuan, Jun; Xiao, Wen-Jing (9 July 2012). "Visible-Light Photoredox Catalysis". Angewandte Chemie International Edition. 51 (28): 6828–6838. doi:10.1002/anie.201200223.
- ^ Fagnoni, Maurizio; Dondi, Daniele; Ravelli, Davide; Albini, Angelo (June 2007). "Photocatalysis for the Formation of the C−C Bond". Chemické recenze. 107 (6): 2725–2756. doi:10.1021/cr068352x. PMID 17530909.
- ^ Romero, Nathan A.; Nicewicz, David A. (10 June 2016). "Organic Photoredox Catalysis". Chemické recenze. 2016 (116): 10075–10166. doi:10.1021/acs.chemrev.6b00057. PMID 27285582.
- ^ Hamada, Taisuke; Ishida, Hitoshi; Usui, Satoshi; Watanabe, Yoshiro; Tsumura, Kazunori; Ohkubo, Katsutoshi (1993). "A novel photocatalytic asymmetric synthesis of (R)-(+)-1,1?-bi-2-naphthol derivatives by oxidative coupling of 3-substituted-2-naphthol with ?-[Ru(menbpy)3]2+[menbpy = 4,4?-di(1R,2S,5R)-(?)-menthoxycarbonyl-2,2?-bipyridine], which possesses molecular helicity". Journal of the Chemical Society, Chemical Communications (11): 909. doi:10.1039/C39930000909.
- ^ Rono, Lydia J.; Yayla, Hatice G.; Wang, David Y.; Armstrong M, ichael F.; Knowles, Robert R. (27 November 2013). "Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization". Journal of the American Chemical Society. 135 (47): 17735–17738. doi:10.1021/ja4100595. PMID 24215561.
- ^ Jones, Wayne E.; Fox, Marye Anne (May 1994). "Determination of Excited-State Redox Potentials by Phase-Modulated Voltammetry". The Journal of Physical Chemistry. 98 (19): 5095–5099. doi:10.1021/j100070a025.
- ^ "Electrochemical Series of Photocatalysts and Common Organic Compounds" (PDF). Merck. Citováno 15. dubna 2019.
- ^ A b Tucker, Joseph W.; Stephenson, Corey R. J. (2012). "Shining Light on Photoredox Catalysis: Theory and Synthetic Applications". The Journal of Organic Chemistry. 77 (4): 1617–1622. doi:10.1021/jo202538x. PMID 22283525.
- ^ Hedstrand, David M.; Kruizinga, Wim H.; Kellogg, Richard M. (January 1978). "Light induced and dye accelerated reductions of phenacyl onium salts by 1,4-dihydropyridines". Čtyřstěn dopisy. 19 (14): 1255–1258. doi:10.1016/S0040-4039(01)94515-0.
- ^ Willner, Itamar; Tsfania, Tamar; Eichen, Yoav (April 1990). "Photocatalyzed and electrocatalyzed reduction of vicinal dibromides and activated ketones using ruthenium(I) tris(bipyridine) as electron-transfer mediator". The Journal of Organic Chemistry. 55 (9): 2656–2662. doi:10.1021/jo00296a023.
- ^ Hironaka, Katsuhiko; Fukuzumi, Shunichi; Tanaka, Toshio (1984). "Tris(bipyridyl)ruthenium(II)-photosensitized reaction of 1-benzyl-1,4-dihydronicotinamide with benzyl bromide". Journal of the Chemical Society, Perkin Transactions 2 (10): 1705. doi:10.1039/P29840001705.
- ^ Kern, Jean-Marc; Sauvage, Jean-Pierre (1987). "Photoassisted C?C coupling via electron transfer to benzylic halides by a bis(di-imine) copper(I) complex". Journal of the Chemical Society, Chemical Communications (8): 546. doi:10.1039/C39870000546.
- ^ Fukuzumi, Shunichi.; Mochizuki, Seiji.; Tanaka, Toshio. (Leden 1990). "Photocatalytic reduction of phenacyl halides by 9,10-dihydro-10-methylacridine: control between the reductive and oxidative quenching pathways of tris(bipyridine)ruthenium complex utilizing an acid catalysis". The Journal of Physical Chemistry. 94 (2): 722–726. doi:10.1021/j100365a039.
- ^ Narayanam, Jagan M. R.; Joseph W. Tucker; Corey R. J. Stephenson (June 5, 2009). "Electron-Transfer Photoredox Catalysis: Development of a Tin-Free Reductive Dehalogenation Procedure". JACS. 131 (25): 8756–8757. doi:10.1021/ja9033582. PMID 19552447.
- ^ Nguyen, John D.; D'Amato, Erica M.; Narayanam, Jagan M. R.; Stephenson, Corey R. J. (2012). "Engaging unactivated alkyl, alkenyl and aryl iodides in visible-light-mediated free radical reactions". Přírodní chemie. 4 (10): 854–859. doi:10.1038/nchem.1452. PMID 23001000.
- ^ Tucker, Joseph W.; Nguyen, John D.; Narayanam, Jagan M. R.; Krabbe, Scott W.; Stephenson, Corey R. J. (28 May 2010). "Tin-free radical cyclization reactions initiated by visible light photoredox catalysis". Chemická komunikace. 46 (27): 4985–4987. doi:10.1039/c0cc00981d. PMID 20512181.
- ^ Furst, Laura; Narayanam, Jagan M. R.; Stephenson, Corey R. J. (4 October 2011). "Total Synthesis of (+)-Gliocladin C Enabled by Visible-Light Photoredox Catalysis". Angewandte Chemie International Edition. 50 (41): 9655–9659. doi:10.1002/anie.201103145. PMC 3496252. PMID 21751318.
- ^ Condie, Allison G.; González-Gómez, José C.; Stephenson, Corey R. J. (10 February 2010). "Visible-Light Photoredox Catalysis: Aza-Henry Reactions via C−H Functionalization". Journal of the American Chemical Society. 132 (5): 1464–1465. doi:10.1021/ja909145y. PMID 20070079.
- ^ Rueping, Magnus; Zhu, Shaoqun; Koenigs, René M. (2011). "Visible-light photoredox catalyzed oxidative Strecker reaction". Chemická komunikace. 47 (47): 12709–11. doi:10.1039/C1CC15643H. PMID 22041859.
- ^ Zhao, Guolei; Yang, Chao; Guo, Lin; Sun, Hongnan; Chen, Chao; Xia, Wujiong (2012). "Visible light-induced oxidative coupling reaction: easy access to Mannich-type products". Chemická komunikace. 48 (17): 2337–9. doi:10.1039/C2CC17130A. PMID 22252544.
- ^ Rueping, Magnus; Zhu, Shaoqun; Koenigs, René M. (2011). "Photoredox catalyzed C–P bond forming reactions—visible light mediated oxidative phosphonylations of amines". Chemická komunikace. 47 (30): 8679–81. doi:10.1039/C1CC12907D. PMID 21720622.
- ^ Freeman, David B .; Furst, Laura; Condie, Allison G.; Stephenson, Corey R. J. (6 January 2012). "Functionally Diverse Nucleophilic Trapping of Iminium Intermediates Generated Utilizing Visible Light". Organické dopisy. 14 (1): 94–97. doi:10.1021/ol202883v. PMC 3253246. PMID 22148974.
- ^ Rueping, Magnus; Koenigs, René M.; Poscharny, Konstantin; Fabry, David C.; Leonori, Daniele; Vila, Carlos (23 April 2012). "Dual Catalysis: Combination of Photocatalytic Aerobic Oxidation and Metal Catalyzed Alkynylation Reactions-C≡C Bond Formation Using Visible Light". Chemistry: A European Journal. 18 (17): 5170–5174. doi:10.1002/chem.201200050.
- ^ Pan, Yuanhang; Wang, Shuai; Kee, Choon Wee; Dubuisson, Emilie; Yang, Yuanyong; Loh, Kian Ping; Tan, Choon-Hong (2011). "Graphene oxide and Rose Bengal: oxidative C–H functionalisation of tertiary amines using visible light". Zelená chemie. 13 (12): 3341. doi:10.1039/C1GC15865A.
- ^ Fu, Weijun; Guo, Wenbo; Zou, Guanglong; Xu, Chen (August 2012). "Selective trifluoromethylation and alkynylation of tetrahydroisoquinolines using visible light irradiation by Rose Bengal". Journal of Fluorine Chemistry. 140: 88–94. doi:10.1016/j.jfluchem.2012.05.009.
- ^ Hari, Durga Prasad; König, Burkhard (5 August 2011). "Eosin Y Catalyzed Visible Light Oxidative C–C and C–P bond Formation". Organické dopisy. 13 (15): 3852–3855. doi:10.1021/ol201376v. PMID 21744842.
- ^ DiRocco, Daniel A.; Rovis, Tomislav (16 May 2012). "Catalytic Asymmetric α-Acylation of Tertiary Amines Mediated by a Dual Catalysis Mode: N-Heterocyclic Carbene and Photoredox Catalysis". Journal of the American Chemical Society. 134 (19): 8094–8097. doi:10.1021/ja3030164. PMC 3354013. PMID 22548244.
- ^ Tucker, Joseph W.; Narayanam, Jagan M. R.; Shah, Pinkey S.; Stephenson, Corey R. J. (2011). "Oxidative photoredox catalysis: mild and selective deprotection of PMB ethers mediated by visible light". Chemická komunikace. 47 (17): 5040–5042. doi:10.1039/c1cc10827a. PMID 21431223.
- ^ Ischay, Michael A .; Anzovino, Mary E.; Du, Juana; Yoon, Tehshik P. (October 2008). "Efficient Visible Light Photocatalysis of [2+2] Enone Cycloadditions". Journal of the American Chemical Society. 130 (39): 12886–12887. doi:10.1021/ja805387f. PMID 18767798.
- ^ Du, Juana; Yoon, Tehshik P. (21 October 2009). "Crossed Intermolecular [2+2] Cycloadditions of Acyclic Enones via Visible Light Photocatalysis". Journal of the American Chemical Society. 131 (41): 14604–14605. doi:10.1021/ja903732v. PMC 2761970. PMID 19473018.
- ^ Ischay, Michael A .; Lu, Zhan; Yoon, Tehshik P. (30 June 2010). "[2+2] Cycloadditions by Oxidative Visible Light Photocatalysis". Journal of the American Chemical Society. 132 (25): 8572–8574. doi:10.1021/ja103934y. PMC 2892825. PMID 20527886.
- ^ Tyson, Elizabeth L.; Farney, Elliot P.; Yoon, Tehshik P. (17 February 2012). "Photocatalytic [2 + 2] Cycloadditions of Enones with Cleavable Redox Auxiliaries". Organické dopisy. 14 (4): 1110–1113. doi:10.1021/ol3000298. PMC 3288794. PMID 22320352.
- ^ Ischay, Michael A .; Ament, Michael S.; Yoon, Tehshik P. (2012). "Crossed intermolecular [2 + 2] cycloaddition of styrenes by visible light photocatalysis". Chemická věda. 3 (9): 2807–2811. doi:10.1039/c2sc20658g. PMC 3439822. PMID 22984640.
- ^ Du, Juana; Espelt, Laura Ruiz; Guzei, Ilia A .; Yoon, Tehshik P. (2011). "Photocatalytic reductive cyclizations of enones: Divergent reactivity of photogenerated radical and radical anion intermediates". Chemická věda. 2 (11): 2115–2119. doi:10.1039/c1sc00357g. PMC 3222952. PMID 22121471.
- ^ Zhao, Guolei; Yang, Chao; Guo, Lin; Sun, Hongnan; Lin, Run; Xia, Wujiong (20 July 2012). "Reactivity Insight into Reductive Coupling and Aldol Cyclization of Chalcones by Visible Light Photocatalysis". The Journal of Organic Chemistry. 77 (14): 6302–6306. doi:10.1021/jo300796j. PMID 22731518.
- ^ Riener, Michelle; Nicewicz, David A. (2013). "Synthesis of cyclobutane lignans via an organic single electron oxidant–electron relay system". Chemická věda. 4 (6): 2625. doi:10.1039/c3sc50643f. PMC 3862357. PMID 24349680.
- ^ Hurtley, Anna E.; Cismesia, Megan A.; Ischay, Michael A .; Yoon, Tehshik P. (June 2011). "Visible light photocatalysis of radical anion hetero-Diels–Alder cycloadditions". Čtyřstěn. 67 (24): 4442–4448. doi:10.1016/j.tet.2011.02.066. PMC 3110713. PMID 21666769.
- ^ A b Lin, Shishi; Ischay, Michael A .; Fry, Charles G.; Yoon, Tehshik P. (7 December 2011). "Radical Cation Diels–Alder Cycloadditions by Visible Light Photocatalysis". Journal of the American Chemical Society. 133 (48): 19350–19353. doi:10.1021/ja2093579. PMC 3227774. PMID 22032252.
- ^ Lin, Shishi; Padilla, Christian E.; Ischay, Michael A .; Yoon, Tehshik P. (June 2012). "Visible light photocatalysis of intramolecular radical cation Diels–Alder cycloadditions". Čtyřstěn dopisy. 53 (24): 3073–3076. doi:10.1016/j.tetlet.2012.04.021. PMC 3375996. PMID 22711942.
- ^ Nicewicz, D. A.; MacMillan, D. W. C. (3 October 2008). "Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes". Věda. 322 (5898): 77–80. doi:10.1126/science.1161976. PMC 2723798. PMID 18772399.
- ^ Nagib, David A .; Scott, Mark E .; MacMillan, David W. C. (12 August 2009). „Enantioselektivní α-trifluormethylace aldehydů pomocí organokatalýzy Photoredox“. Journal of the American Chemical Society. 131 (31): 10875–10877. doi:10.1021 / ja9053338. PMC 3310169. PMID 19722670.
- ^ Shih, Hui-Wen; Vander Wal, Mark N.; Grange, Rebecca L.; MacMillan, David W. C. (6 October 2010). "Enantioselective α-Benzylation of Aldehydes via Photoredox Organocatalysis". Journal of the American Chemical Society. 132 (39): 13600–13603. doi:10.1021/ja106593m. PMC 3056320. PMID 20831195.
- ^ Neumann, Matthias; Füldner, Stefan; König, Burkhard; Zeitler, Kirsten (24 January 2011). „Nekovová kooperativní asymetrická katalýza organophotoredoxu s viditelným světlem“. Angewandte Chemie International Edition. 50 (4): 951–954. doi:10,1002 / anie.201002992. PMID 20878819.
- ^ Pirnot, M. T.; Rankic, D. A.; Martin, D. B. C.; MacMillan, D. W. C. (28 March 2013). "Photoredox Activation for the Direct -Arylation of Ketones and Aldehydes". Věda. 339 (6127): 1593–1596. doi:10.1126/science.1232993. PMC 3723331. PMID 23539600.
- ^ Zhang, Shi-Lei; Xie, He-Xin; Zhu, Jin; Li, Hao; Zhang, Xin-Shuai; Li, Jian; Wang, Wei (1 March 2011). "Organocatalytic enantioselective β-functionalization of aldehydes by oxidation of enamines and their application in cascade reactions". Příroda komunikace. 2: 211. doi:10.1038/ncomms1214. PMID 21364550.
- ^ Petronijević, Filip R.; Nappi, Manuel; MacMillan, David W. C. (22 November 2013). "Direct β-Functionalization of Cyclic Ketones with Aryl Ketones via the Merger of Photoredox and Organocatalysis". Journal of the American Chemical Society. 135 (49): 131122154626007. doi:10.1021/ja410478a. PMC 3934322. PMID 24237366.
- ^ Barton, Derek H.R.; Csiba, Maria A.; Jaszberenyi, Joseph Cs. (Květen 1994). "Ru(bpy)32+-mediated addition of Se-phenyl p-tolueneselenosulfonate to electron rich olefins". Čtyřstěn dopisy. 35 (18): 2869–2872. doi:10.1016/S0040-4039(00)76646-9.
- ^ Yasu, Yusuke; Koike, Takashi; Akita, Munetaka (17 September 2012). "Three-component Oxytrifluoromethylation of Alkenes: Highly Efficient and Regioselective Difunctionalization of C=C Bonds Mediated by Photoredox Catalysts". Angewandte Chemie International Edition. 51 (38): 9567–9571. doi:10.1002/anie.201205071. PMID 22936394.
- ^ Yasu, Yusuke; Koike, Takashi; Akita, Munetaka (3 May 2013). "Intermolecular Aminotrifluoromethylation of Alkenes by Visible-Light-Driven Photoredox Catalysis". Organické dopisy. 15 (9): 2136–2139. doi:10.1021/ol4006272. PMID 23600821.
- ^ Wilger, Dale J.; Gesmundo, Nathan J.; Nicewicz, David A. (2013). "Catalytic hydrotrifluoromethylation of styrenes and unactivated aliphatic alkenes via an organic photoredox system". Chemická věda. 4 (8): 3160. doi:10.1039/c3sc51209f.
- ^ Hamilton, David S.; Nicewicz, David A. (14 November 2012). "Direct Catalytic Anti-Markovnikov Hydroetherification of Alkenols". Journal of the American Chemical Society. 134 (45): 18577–18580. doi:10.1021/ja309635w. PMC 3513336. PMID 23113557.
- ^ Nguyen, Tien M .; Nicewicz, David A. (3 July 2013). "Anti-Markovnikov Hydroamination of Alkenes Catalyzed by an Organic Photoredox System". Journal of the American Chemical Society. 135 (26): 9588–9591. doi:10.1021/ja4031616. PMC 3754854. PMID 23768239.
- ^ Grandjean, Jean-Marc M.; Nicewicz, David A. (2 April 2013). "Synthesis of Highly Substituted Tetrahydrofurans by Catalytic Polar-Radical-Crossover Cycloadditions of Alkenes and Alkenols". Angewandte Chemie International Edition. 52 (14): 3967–3971. doi:10.1002/anie.201210111. PMID 23440762.
- ^ Perkowski, Andrew J.; Nicewicz, David A. (17 July 2013). "Direct Catalytic Anti-Markovnikov Addition of Carboxylic Acids to Alkenes". Journal of the American Chemical Society. 135 (28): 10334–10337. doi:10.1021/ja4057294. PMC 3757928. PMID 23808532.