Nemoc BENTA - BENTA disease
| Nemoc BENTA | |
|---|---|
| Ostatní jména | Expanze B-buněk s NF-kB a T-buněčnou anergií |
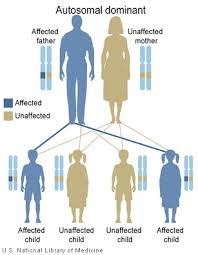 | |
| U každého dítěte, které má postižený rodič, existuje 50% pravděpodobnost přenosu mutace bez ohledu na pohlaví dítěte autozomálně dominantní | |
| Specialita | Imunologie |
Nemoc BENTA je vzácný genetická porucha z imunitní systém. BENTA znamená „B buňka expanze s NF-kB a T buňka anergie "a je způsobena zárodečnou linií heterozygotní zisk funkce mutace v gen KARTA 11 (viz položka OMIM č. 607210 ). Tato porucha je charakterizována polyklonálními B buňkami lymfocytóza s počátkem kojeneckého věku, splenomegalie, lymfadenopatie, mírné imunodeficience a zvýšené riziko lymfom. Vyšetřovatelé Andrew L. Snow a Michael J. Lenardo u Národní institut pro alergie a infekční choroby v USA National Institutes of Health poprvé charakterizovala nemoc BENTA v roce 2012. Současná laboratoř Dr. Snowa na Uniformed Services University of Health Sciences nyní tuto poruchu aktivně studuje.[1][2]
Prezentace
Jedinci s onemocněním BENTA mají polyklonální B buňka lymfocytóza (tj. přebytek B buněk) vyvíjející se v dětství, kromě splenomegalie a lymfadenopatie. Pacienti mohou mít nízkou hladinu sérum IgM a mírně anergický T buňky. Tyto funkce pravděpodobně přispívají k mírnému imunodeficience při onemocnění BENTA. Pacienti jsou obecně náchylní k rekurenci sinopulmonální a ušní infekce v dětství a mohou být náchylnější k určitým virům, včetně Virus Epstein-Barr, BK virus, a molluscum contagiosum.[1]
Genetika

Onemocnění BENTA je způsobeno zárodečná linie -kódováno mutace zesílení funkce v genu CARD11. Toto je gen 138 kB mapovaný na chromozom 7p22 s 26 exony kódující 1 154 aminokyselinový protein.[3][4] Protein CARD11 (také známý jako CARMA1) je a lešení protein potřebné pro aktivaci NF-kB v B i T lymfocytech. Mutace zesílení funkce řídí konstitutivní aktivaci NF-kB v obou typech buněk. Většina mutací je lokalizována v doméně coiled-coil (exony 4-9) proteinu nebo jen proti ní. Fenotypy pacientů to také naznačují Diferenciace B buněk může být u BENTA nemoci částečně narušena, což přispívá k nízkému procentu B-buněk přepínaných do třídy a paměti.[1][2]
Mutace germinálního zesílení funkce na CARD11 projevují méně závažné onemocnění než mutace ztráty funkce při nedostatku karty CARD11 (OMIM # 615206 ), an autozomálně recesivní stav projevující se v závažná kombinovaná imunodeficience.[1]
Mutace CARD11 se ziskem funkce spojené s onemocněním BENTA mohou také předisponovat pacienty k B buňkám malignita. Důležité je, že hyperaktivní NF-κB je často spojován s B buněčná malignita a konkrétně somatický mutace CARD11 se ziskem funkce se často vyskytují u difuzního velkobuněčného B-lymfomu (DLBCL ). Většina pacientů s BENTA se však vyskytuje polyklonální Akumulace B buněk bez známek oligoklonálních nebo monoklonálních populací (tj. Malignity). Nezdá se, že by tyto mutace byly spojeny s Malignity T buněk.[2]
Dědictví
Tato porucha se dědí v autosomálně dominantní způsob. Autosomální odkazuje na skutečnost, že každý člověk má dvě KARTU 11 alely, jeden zděděný od každého rodiče. To je v rozporu s spojeno se sexem chromozomy. Dominantní znamená, že abnormální alela dominuje shodné normální alele. Pouze jedna ze dvou kopií (alel) KARTY 11 musí být abnormální, aby osoba mohla mít onemocnění BENTA. Onemocnění BENTA může také nastat spontánně u pacienta v důsledku a de novo mutace v KARTĚ 11, což znamená, že mutace nebyla zděděna od rodičů. V tomto případě by pacient mohl mutaci přenést na své děti.
Děti rodiče, který nese mutaci CARD11, mají 50% šanci zdědit mutaci. V rodině je riziko, že každé dítě zdědí mutovanou alelu CARD11, nezávislé na tom, zda mutaci zdědili ostatní sourozenci. Například pokud mají první čtyři děti v rodině mutaci, další dítě má stejné 50% riziko zdědění mutace. U dětí, které nezdědí mutaci, se nevyvinete onemocnění BENTA ani jej nepřenáší na své děti.
Diagnóza
Většina pacientů mononukleární buňky periferní krve jsou polyklonální naivní zralé B buňky, s výrazným nárůstem nezralých, přechodná B buňka čísla (označená jako CD10 +).[5] Procenta cirkulujících třídních a paměťových B buněk jsou velmi nízká a in vitro studie ukazují špatnou diferenciaci B buněk a sekreci imunoglobulinu. Sérové IgM je u většiny pacientů nízké, zatímco celkové IgG a IgA mohou být na spodní hranici normálu. Pacienti vykazují defektní produkci protilátek proti nezávislosti na T buňkách, vakcíny na bázi polysacharidů. Někteří pacienti nemusí stanovit ochranné titry protilátek na jiné vakcíny, jako je např spalničky a varicella zoster virus.[2][6]
Počty T buněk jsou obecně v normálním rozmezí nebo těsně nad ním. In vitro stimulace T buněk ukazuje, že jak CD4 +, tak CD8 + T buňky jsou méně citlivé než normální, což naznačuje mírné T buňky anergie u pacientů.[1]
Diagnóza leukémie lze u těchto pacientů obecně vyloučit na základě neobvyklého vzhledu malého odpočinku lymfocyty v krvi; pacienti však musí být pečlivě sledováni s ohledem na jakékoli příznaky monoklonální nebo oligoklonální expanze B buněk, protože může existovat zvýšené riziko malignity B buněk. Konkrétně byl hlášen vývoj u jednoho pacienta s onemocněním BENTA B lymfocytární chronická lymfocytární leukémie (B-CLL) jako dospělý.[1]
Léčba
V současné době existuje minimální terapeutická intervence pro onemocnění BENTA. Pacienti jsou pečlivě sledováni na přítomnost infekcí a na známky monoklonální nebo oligoklonální expanze B buněk, které by mohly naznačovat malignitu B buněk. Splenektomie je nepravděpodobné, že sníží zátěž B buněk; počty B buněk v periferní krvi významně vzrostly u tří pacientů, kteří podstoupili tento zákrok. Zbývá určit, zda imunosupresivní léky, včetně léků snižujících hladinu B buněk, jako jsou rituximab, by mohly být účinné při léčbě choroby BENTA.[1]
Reference
- ^ A b C d E F G Turvey, SE; Durandy, A; Fischer, A; Fung, SY; Geha, RS; Gewies, A; Giese, T; Greil, J; Keller, B; McKinnon, ML; Neven, B; Rozmus, J; Ruland, J; Sníh, AL; Stepensky, P; Warnatz, K (2014). „Komplex signalosomů CARD11-BCL10-MALT1 (CBM): Vstupujeme do záře reflektorů primární imunodeficience člověka“. The Journal of Allergy and Clinical Immunology. 134 (2): 276–84. doi:10.1016 / j.jaci.2014.06.015. PMC 4167767. PMID 25087226.
- ^ A b C d E Snow, A. L .; Xiao, W .; Stinson, J. R .; Lu, W .; Chaigne-Delalande, B .; Zheng, L .; Pittaluga, S .; Matthews, H. F .; Schmitz, R .; Jhavar, S .; Kuchen, S .; Kardava, L .; Wang, W .; Lamborn, I. T .; Jing, H .; Raffeld, M .; Moir, S .; Fleisher, T. A .; Staudt, L. M .; Su, H. C .; Lenardo, M. J. (5. listopadu 2012). „Vrozená lymfocytóza B buněk vysvětlena novými mutacemi zárodečné linie CARD11“. Journal of Experimental Medicine. 209 (12): 2247–2261. doi:10.1084 / jem.20120831. PMC 3501355. PMID 23129749.
- ^ "Rodina domén náboru kaspáz CARD11, člen 11 [Homo sapiens (člověk)]" ". NCBI> Geny a exprese> Gen. NCBI. Citováno 4. září 2014.
- ^ „Protein obsahující nábor domén Caspase 11 [Homo sapiens]“. NCBI. Citováno 4. září 2014.
- ^ Chung JB1, Silverman M, Monroe JG. (Červen 2003). "Přechodné B buňky: krok za krokem k imunitní kompetenci". Trends Immunol. 24 (6): 343–9. PMID 12810111.CS1 maint: používá parametr autoři (odkaz)
- ^ Kniffin, Cassandra. „# 606445 Trvalá polyklonální lymfocytóza B-buněk; PPBL“. OMIM. Univerzita Johna Hopkinse. Archivovány od originál dne 24. září 2015. Citováno 4. září 2014.
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |