G & T-sekv - G&T-Seq - Wikipedia
G & T-seq (zkratka pro sekvenování jednobuněčného genomu a transkriptomu) je nová forma sekvenování jedné buňky technika umožňující jednomu současně získat obojí transkriptomický a genomický data z jednotlivých buněk, což umožňuje přímé srovnání údajů o genové expresi s odpovídajícími genomickými údaji ve stejné buňce ...[1]
Pozadí
Nástup sekvenování jednotlivých buněk poskytl vědcům nástroje k vyřešení genotypicky a fenotypicky odlišné buňky ve smíšené populaci.[2] V případech, kdy takové heterogenita je relevantní, například u nádorů, tato technika umožňuje studium klonální vztahy a vývoj nádoru.[3] Rovněž lze podrobněji studovat vzácné typy buněk a vzorky, které jinak obsahují nízký počet buněk, například v případě cirkulujících nádorových buněk.[4] Nicméně předchozí metody příprava knihovny typicky zahrnují zachycení buď mRNA nebo genomové DNA (gDNA), ale ne obojí.[5] Současným zachycením a sekvenováním DNA i RNA metodou nazvanou G&T sekvenování jsou vědci schopni získat informace o sekvencích pro analýzu genomu i transkriptomu z knihoven jednotlivých buněk, což umožňuje integrované studie zahrnující obě sítě. Jako důkaz konceptu autoři G & T-seq prokázali svou schopnost získat jak messenger RNA (mRNA), tak genomovou DNA (gDNA) pomocí paramagnetické korálky s biotinylovaný oligo-deoxy-thyminový (dT) primer oddělit polyadenylovanou (Poly-A) RNA od její gDNA před amplifikací a přípravou knihovny. Ukazují to validační experimenty na G & T-sekvenci prováděné pomocí buněčných linií s předchozími dostupnými sekvenčními daty sekvenční pokrytí, profil genové exprese, a Profily počtu kopií DNA byly spolehlivě reprodukovány sekvenováním G&T a že tato metoda byla schopna volat většinu (87%) všech dříve anotovaných variant jednotlivých nukleotidů (SNV) v těchto buněčných liniích. Autoři na tomto základě tvrdili, že proces fyzikální separace mRNA od gDNA neovlivnil negativně výtěžek ani kvalitu sekvenčních dat.[1]
Metody
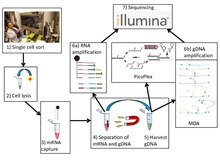
Podobně jako u běžného jednobuněčného sekvenování zahrnuje G & T-seq sběr a lýzu požadovaných buněk. Nicméně gDNA i polyA-mRNA jsou zachyceny a fyzicky odděleny před amplifikací a konstrukcí knihovny pro analýzu pomocí sekvenční platformy.
Separace polyadenylované RNA z genomové DNA
Sekvenování G&T odděluje mRNA od gDNA pomocí dříve popsaného nezaujatého globálního postupu amplifikace.[6] Nejprve se mRNA izoluje na specializovaných oligo-dT (5'-biotin-triethylenglykol-AAGCAGTGGTATCAACGCAGAGTAC (T) 30VN-3 ') konjugovaných na paramagnetické kuličky spojené se streptavidinem.[7] Oligo-dT se váže na poly-A ocasy zpracované mRNA a loví je ze zásoby genomového materiálu. Poté jsou paramagnetické kuličky prostorově izolovány magnetizací. Genomický materiál zbývající v supernatant je extrahován a fyzicky oddělen od mRNA.[1]
Zesílení a sekvenování
Autoři, kteří vyvinuli G & T-seq, využili a ověřili dvě metody pro amplifikaci celého genomu: amplifikace s vícenásobným posunem a PicoPlex. Jiné metody, jako např MALBAC, může být použitelné, ale ještě musí být ověřeno.[1][8]
Zesílení vícenásobného posunutí
MDA amplifikační techniku lze použít ke generování dlouhých a vysoce kvalitních čtení, která produkují sekvenční data srovnatelné kvality s hromadným sekvenováním pomocí Amplifikace PCR.[9] Tato metoda zahrnuje použití hexamerových primerů, které se náhodně vážou na templát, následované prodloužením DNA pomocí phi29 DNA polymeráza. Po dosažení 5 'konce následného primeru polymeráza vytlačí tento protahovací řetězec, aby pokračovala v syntéze. Posunuté vlákno se otevře pro párování s více primery, což umožňuje zesílení posunutého vlákna. Proces pokračuje a produkuje rozvětvenou knihovnu DNA, kterou lze rozřezat a sekvenovat. Autoři techniky G&T zjistili, že ačkoli MDA použitý v G & T-seq přinesl genomové pokrytí podobné šíře jako MDA prováděné v konvenčním sekvenování jednotlivých buněk, distribuce pokrytí čtením byla v celém genomu méně rovnoměrná.[1]
PicoPlex
Ačkoli MDA produkuje kvalitnější čtení vhodná pro SNP analýzu, profily počtu kopií DNA generované takovou technikou nejsou vysoce přesné a reprodukovatelné kvůli jeho nejednotné amplifikaci.[5][10] Ukázalo se, že alternativní technika zvaná PicoPlex, vyvinutá společností Rubicon Genomics, přináší lepší výsledky.[1] Zde došlo k ligaci prodloužení náhodných primerů k adaptér vytváří komplementární vlákno s adaptérem, který po denaturaci a náhodném přepracování vytvoří dvouvláknový fragment s doplňkovými adaptéry. Denaturace na jednotlivé prameny umožňuje vznik vlásenky vzhledem k komplementární povaze jejich adaptérů, vytvoření knihovny vlásenkové smyčky, kterou nelze použít pro následnou amplifikaci, čímž se zabrání exponenciálnímu zesílení počátečního zkreslení.[11][12]
amplifikace cDNA
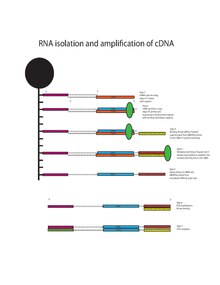
Messenger RNA navázaná na oligo-dT je přepsána reverzně cDNA za použití oligo-dT primerů s přídavkem templátu přepínajícího oligo (TSO, 5 "-AAGCAGTGGTATCAACGCAGAGTACrGrG + G-3 ') a reverzní transkriptázy Superscript II.[13][14] Reverzní transkriptáza Superscript II má další aktivitu terminální transferázy, která přidává variabilní počet cytosinových zbytků na konec 3 'koncové molekuly cDNA. Přesah 3 'cytosinových zbytků se váže na TSO a vytváří rozšířenou šablonu. Reverzní transkriptáza Superscript II přepíná šablony a pokračuje v přepisu, aby dokončil 3 'konec cDNA. To má za následek úplnou cDNA obsahující 5 'oligo-dT primer, cDNA transkribovanou z mRNA a 3' univerzální primární místo pro syntézu druhého řetězce. CDNA prochází amplifikací pomocí univerzálního primeru (5'-AAGCAGTGGTATCAACGCAGAGT-3 ' ) po dobu 18 cyklů PCR, než podstoupí přípravu knihovny pomocí soupravy Nextera XT Kit od společnosti Illumina a sekvenování platformou Illumina HiSeq.[1][15]
Alternativní techniky
Podobnou metodou jako G & T-seq, vyvinutou před několika měsíci, je DR-seq (sekvenování DNA a RNA). Primárním rozdílem mezi těmito dvěma technikami je krok amplifikace, kdy dochází k amplifikaci DNA a polyA-RNA bez jejich předchozí separace.[16] DR-seq používá náhodné primování, kde se primery obsahující běžnou 27-nukleotidovou sekvenci spolu s variabilním 8-nukleotidem (primery ad2) vážou na různá místa na cDNA.[12] Přestože na většině cDNA existuje více (50–250) vazebných míst pro primer, každý originál (tj. Není produktem amplifikace / in vitro transkripce ) Molekula cDNA je obvykle aktivována pouze jednou během počátečního amplifikačního kroku, čímž se vytvoří jediná amplikon unikátní délky, obsahující na 5 'konci primer ad2. 3 'konec obsahuje primer ad1, což je původní primer poly-dT používaný pro reverzní transkripci. Tento jedinečný amplikon se nazývá identifikátor založený na délce. Důležité je, že identifikátor založený na délce je vytvořen, ale není amplifikován tímto kvazilineárním krokem PCR. Počet jedinečných identifikátorů založených na délce každého genu lze poté použít k odvození počtu původních molekul cDNA (a tedy mRNA) přítomných pro gen, což poskytuje metodu odhadu genové exprese, která se vyhne účinku zkreslení amplifikace. K další amplifikaci cDNA pro RNA-seq procházejí cDNA amplikony generované původním krokem PCR transkripcí in vitro s použitím promotoru T7 zabudovaného do primeru ad1, aby se zajistilo, že transkripty RNA pocházejí z cDNA, nikoli z gDNA.
Mezi výhody techniky DR-seq patří snížení možnosti kontaminace a ztráty RNA, protože je vynechán další krok separace DNA / RNA. Rovněž je zkreslení amplifikace sníženo v důsledku použití výše uvedených identifikátorů založených na délce. Protože však DNA a polyA-RNA nejsou separovány před amplifikací a následným sekvenováním, exonální oblasti musí být výpočetně maskovaný, aby bylo možné určit počet kopií, ponechat pouze čtení, která pocházejí z gDNA. To vytváří problémy pro přesné stanovení počtu kopií z gDNA. Autoři poznamenávají, že počet kopií ve velkých genomických oblastech není zřejmě ovlivněn maskováním, protože kódující oblasti tvoří relativně malou část genomu.[16]
Aplikace
Sekvenování duálního genomu a transkriptomu umožňuje vědcům stanovit korelace genomových aberací s vysokým rozlišením se změnami úrovní transkripce. Například autoři této techniky dokázali detekovat jednotlivé buňky chromozomálně aneuploidie, a stanovit, že tyto aneuploidie odpovídaly zvýšené nebo snížené celkové expresi chromozomálních genů, když došlo k příslušnému chromozomálnímu zisku (např. Trizomie ) nebo ztráta. Subchromozomální změny lze také korelovat se změnami v expresi genů v postižených lokusech. Autoři také dokázali najít transkripci fúze a lokalizovat zlom chromozomů ve stejné buňce, což vedlo k fúzi.[1]
G & T-seq také poskytuje strategii pro vytváření kauzálních vazeb mezi asociacemi genotypu a fenotypu v jednotlivých buňkách (např. Nekódující SNV). Zatímco hromadné sekvenování genomu a transkriptomu může člověku umožnit spojit soubor genotypových znaků s průměrnými expresními vzory v populaci buněk, přehlíží jemné nebo časové rozdíly mezi jednotlivými buňkami, které mohou vzniknout v důsledku buněčné ekologie.[17] To představuje překážku pro výzkumné pracovníky, kteří se snaží přesně určit genomické příčiny základních transkripčních změn, zejména při kombinaci s nádorovými vzorky, kde je rozšířená heterogenita a genetické variace na pozadí by mohly zmást relevantní mutace.[3][18][19] Konvenční sekvenování jednotlivých buněk na druhé straně brání tomu, aby člověk vytvořil přímou asociaci mezi mutacemi a změnami v transkriptomu, protože v procesu dochází ke ztrátě DNA nebo RNA. Vědci by tradičně museli používat jiné metody, jako je klasifikace na základě buněčných markerů. Tyto metody diskriminace se však spoléhají na dostupnost specifických protilátek a poskytují relativně hrubou diskriminaci ve srovnání se sekvenováním, protože exprese markerů buněčného povrchu tvoří pouze zlomek jejího celkového fenotypu[20][21]
Nakonec separace DNA od RNA připravuje cestu pro duální sekvenování epigenomu a transkriptomu, dvou složek buňky, které jsou navzájem složitě spojeny. To by však vyžadovalo ověření pomocí konvenčních jednotlivých buněk bisulfitové sekvenování aby se zajistilo, že separace DNA a RNA neovlivní Methylace DNA postavení.
Úvahy
Zkreslení GC
Amplifikace MDA má inherentní zkreslení proti opakujícím se sekvencím, které byly v produktech MDA nedostatečně zastoupeny. V kontextu sekvenování G&T to má za následek snížený počet čtení jako% z Obsah GC zvýšení pro konkrétní region.
Distribuce pokrytí čtení
Porovnání amplifikace reziduální genomové DNA jedné buňky po izolaci mRNA pomocí MDA s amplifikací genomu jediné DNA bez izolace mRNA pomocí MDA ukázalo méně rovnoměrně distribuované pokrytí v genomu po izolaci mRNA. Ačkoli došlo ke snížení distribuce pokrytí, nebylo to z velké části.
Vyloučení alternativní RNA
Popsaná izolace mRNA popsanou technikou G & T-seq je schopna zachytit pouze mRNA, které mají dostatečně dlouhý ocas poly-A, který může být zachycen návnadou oligo-dT.[6] Toto není úplná reprezentace mRNA přítomné v buňce. Některé mRNA mají zásadní roli ve fenotypové expresi, ale nepředstavují standardní délku polyA ocasu kvůli alternativní polyadenylaci.[22] G& Ts srovnání korelace genotyp-fenotyp tedy nemusí nutně představovat nejlepší příčinnou souvislost mezi těmito dvěma.
Korelace exprese proteinů
Izolace mRNA není jedinou překážkou při vytváření vztahu genotyp-fenotyp. Nestačí použít mRNA jako náhradu k celkové expresi proteinu, protože existují i jiné druhy RNA, které také hrají důležitou roli ve fenotypové expresi. Další pomocnou technikou, která může posílit tvrzení sekvenování G&T, je celková analýza proteomu hmotnostní spektrometrií, která poskytuje lepší prezentaci vztahu mezi genomovými změnami a fenotypovou prezentací[15]
Reference
- ^ A b C d E F G h Macaulay, I.C .; Haerty, W .; Kumar, P .; Li, Y. I .; Hu, T. X .; Teng, M. J .; Voet, T. (2015). „G & T-seq: Paralelní sekvenování jednobuněčných genomů a transkriptomů“. Přírodní metody. 12 (6): 519–22. doi:10.1038 / nmeth.3370. PMID 25915121.
- ^ Wang, X. Sekvenování jednotlivých buněk a systémová imunologie (Sv. 5). Springer
- ^ Xi-Xi Chen, Fan Bai (2015). „Jednobuněčné analýzy cirkulujících nádorových buněk“. 癌症 生物学 与 医学 : 英文版. 12 (3): 184–192. doi:10,7497 / j.issn.2095-3941.2015.0056. PMC 4607822. PMID 26487963.
- ^ A b Grün, D .; van Oudenaarden, A. (2015). „Návrh a analýza jednobuněčných sekvenčních experimentů“. Buňka. 163 (4): 799–810. doi:10.1016 / j.cell.2015.10.039. PMID 26544934.
- ^ A b Klein, C. A .; Seidl, S .; Petat-Dutter, K .; Offner, S .; Geigl, J. B .; Schmidt-Kittler, O .; Baeuerle, P. A. (2002). "Kombinovaná transkriptomová a genomová analýza jednotlivých mikrometastatických buněk". Přírodní biotechnologie. 20 (4): 387–92. doi:10.1038 / nbt0402-387. PMID 11923846.
- ^ Picelli, S .; Faridani, O. R .; Björklund, Å. K .; Winberg, G .; Sagasser, S .; Sandberg, R. (2014). "Plná délka RNA-seq z jednotlivých buněk pomocí Smart-seq2". Přírodní protokoly. 9 (1): 171–81. doi:10.1038 / nprot.2014.006. PMID 24385147.
- ^ Chapman, A. R .; On, Z .; Lu, S .; Yong, J .; Tan, L .; Tang, F .; Xie, X. S. (2015). „Zesílení transkriptomu jedné buňky pomocí MALBAC“. PLOS ONE. 10 (3): e0120889. doi:10.1371 / journal.pone.0120889. PMC 4378937. PMID 25822772.
- ^ Blanco, L .; Bernad, A .; Lázaro, J. M .; Martón, G .; Garmendia, C .; Salas, M. (1989). „Vysoce účinná syntéza DNA pomocí fágové fí29 DNA polymerázy. Symetrický způsob replikace DNA“. Journal of Biological Chemistry. 264 (15): 8935–8940. PMID 2498321.
- ^ Voet, T .; Kumar, P .; Van Loo, P .; Cooke, S.L .; Marshall, J .; Lin, M .; Campbell, P. J. (2013). „Sekvenování genomu párovaných párů na jednom konci odhaluje strukturální variace na buněčný cyklus. Výzkum nukleových kyselin. 41 (12): 6119–6138. doi:10.1093 / nar / gkt345. PMC 3695511. PMID 23630320.
- ^ „PGS / PGD.“ Rubikonová genomika. N.p.., n.d. Web. 25. února 2016
- ^ A b Zong, C .; Lu, S .; Chapman, A. R .; Xie, X. S. (2012). "Detekce celého nukleomu jednonukleotidů a variací počtu kopií jedné lidské buňky" (PDF). Věda. 338 (6114): 1622–1626. doi:10.1126 / science.1229164. PMC 3600412. PMID 23258894.
- ^ Goetz, J. J .; Trimarchi, J. M. (2012). "Sekvenování transkriptomů jednotlivých buněk pomocí Smart-Seq". Přírodní biotechnologie. 30 (8): 763–765. doi:10,1038 / nbt.2325. PMID 22871714.
- ^ „Sada pro přípravu knihovny Nextera XT DNA.“ Sada pro přípravu DNA knihovny Nextera XT. N.p., n.d. Web. 25. února 2016.
- ^ A b Maier, T .; Güell, M .; Serrano, L. (2009). "Korelace mRNA a proteinu ve složitých biologických vzorcích". FEBS Dopisy. 583 (24): 3966–3973. doi:10.1016 / j.febslet.2009.10.036. PMID 19850042.
- ^ A b Dey, S. S .; Kester, L .; Spanjaard, B .; Bienko, M .; van Oudenaarden, A. (2015). "Integrované sekvenování genomu a transkriptomu stejné buňky". Přírodní biotechnologie. 33 (3): 285–289. doi:10,1038 / nbt.3129. PMC 4374170. PMID 25599178.
- ^ Shapiro, E .; Biezuner, T .; Linnarsson, S. (2013). „Technologie založené na sekvenování jednotlivých buněk způsobí revoluci ve vědě celého organismu.“ Genetika hodnocení přírody. 14 (9): 618–630. doi:10.1038 / nrg3542. PMID 23897237.
- ^ Xu, X .; Hou, Y .; Yin, X .; Bao, L .; Tang, A .; Song, L .; On, W. (2012). „Jednobuněčné sekvenování exomu odhaluje charakteristiky nukleotidové mutace nádoru ledvin“. Buňka. 148 (5): 886–895. doi:10.1016 / j.cell.2012.02.025. PMID 22385958.
- ^ Patel, A. P .; Tirosh, I .; Trombetta, J. J .; Shalek, A. K .; Gillespie, S. M .; Wakimoto, H .; Louis, D. N. (2014). „Jednobuněčný RNA-seq zdůrazňuje intratumorální heterogenitu v primárním glioblastomu“. Věda. 344 (6190): 1396–1401. doi:10.1126 / science.1254257. PMC 4123637. PMID 24925914.
- ^ Vaughan, Christopher. "Nový způsob třídění buněk bez omezení tradičními metodami." Centrum zpráv. Stanford Medicine, 30. března 2015. Web. 25. února 2016
- ^ Bidlingmaier, S .; Zhu, X .; Liu, B. (2008). „Užitečnost a omezení glykosylovaných lidských epitopů CD133 při definování kmenových buněk rakoviny“. Journal of Molecular Medicine. 86 (9): 1025–1032. doi:10.1007 / s00109-008-0357-8. PMC 2585385. PMID 18535813.
- ^ De Klerk, E .; AC; Hoen, P. (2015). "Alternativní transkripce, zpracování a translace mRNA: poznatky ze sekvenování RNA". Trendy v genetice. 31 (3): 128–139. doi:10.1016 / j.tig.2015.01.001. PMID 25648499.