Hypotrichóza s juvenilní makulární dystrofií - Hypotrichosis with juvenile macular dystrophy - Wikipedia
| Hypotrichóza s juvenilní makulární dystrofií | |
|---|---|
| Ostatní jména | Hypotrichóza s juvenilní makulární degenerací[1] |
 | |
| Hypotrichóza (řídký růst vlasů) u 5letého chlapce s HJMD | |
Hypotrichóza s juvenilní makulární dystrofií (HJMD nebo CDH3) je extrémně vzácný vrozené onemocnění charakterizovaný řídkým růstem vlasů (hypotrichóza ) od narození a progresivní makulární rohovková dystrofie.
Příznaky a symptomy
Růst vlasů na hlavě je znatelně méně plný než obvykle a chloupky jsou velmi slabé; zbytek těla vykazuje normální vlasy. Makulární degenerace přichází pomalu se zhoršením centrálního vidění, což vede ke ztrátě schopnosti čtení. Dotčené osoby se jinak mohou vyvíjet zcela zdravým způsobem; průměrná délka života je normální.[Citace je zapotřebí ]
Způsobit
Hypotrichóza s juvenilní makulární dystrofií je autosomálně recesivní dědičné onemocnění.[2] Je to způsobeno kombinací mutací (složená heterozygotnost ) v CDH3 gen, který kóduje kadherin-3 (také známý jako P-kadherin), a protein vázající vápník za který je zodpovědný buněčná adheze v různých tkáních.[Citace je zapotřebí ]
Diagnóza
Výrazně anomální růst vlasů by měl vést k a vyšetření sítnice nejpozději do vstupu do školy, protože slabé vidění nemusí být při běžných lékařských prohlídkách nutně detekováno. Potvrzení diagnózy, které je nezbytné pro jakékoli budoucí terapeutické možnosti, je možné pouze pomocí a molekulárně genetické diagnóza v kontextu genetické poradenství.[Citace je zapotřebí ]
Vyšetřovací metoda
Rozsah sítnice škoda je hodnocena fluorescenční angiografie, sítnicové skenování a optická koherentní tomografie; elektrofyziologická vyšetření jako např elektroretinografie (ERG) nebo multifokální elektroretinografie Lze také použít (mfERG).[Citace je zapotřebí ]
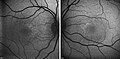
Fluorescenční angiogram pětiletého chlapce s HJMD

Optická koherentní tomografie 5letého chlapce s HJMD

Fundoskopie, levé oko 5letého chlapce s HJMD

Fundoskopie, pravé oko 5letého chlapce s HJMD




Diferenciální diagnostika
Anomálie vlasová šachta zapříčiněno ektodermální dysplázie by mělo být vyloučeno. Mutace v genu CDH3 se mohou také objevit v EEM syndrom.
Léčba
Porucha není léčena. Řada studií se zabývá genová terapie, přeskakování exonu a CRISPR interference nabídnout naději do budoucnosti. Přesné stanovení prostřednictvím potvrzené diagnózy genetické mutace, ke které došlo, nabízí také potenciální přístupy mimo genovou substituci pro konkrétní skupinu, a to v případě diagnózy tzv. nesmyslná mutace mutace, kde a stop kodon je produkován změnou jedné základny v Sekvence DNA. To má za následek předčasné ukončení biosyntéza bílkovin, což má za následek zkrácený a buď nefunkční nebo funkčně narušený protein. V tom, čemu se někdy říká „read-through therapy“, překladový skákání stop kodonu, vedoucí k funkčnímu proteinu, lze vyvolat zavedením specifických látek. Tento přístup je však myslitelný pouze v případě úzce ohraničených mutací, které způsobují různá onemocnění.[Citace je zapotřebí ]
Plánování života
Onemocnění, které ohrožuje zrak a navíc způsobuje vlasovou anomálii, která je zjevná cizím lidem, způsobuje škodu nad rámec fyzické. Není proto divu, že zjištění diagnózy je pro pacienta šokem. To platí stejně pro postižené děti i pro jejich rodiče a příbuzné. Jsou konfrontováni s prohlášením, že v současné době neexistují žádné možnosti léčby. Pravděpodobně se nikdy v životě necítili tak sami a opuštěni. Napadne mě otázka: „Proč já / moje dítě?“ Vždy však existuje naděje, a zvláště u postižených dětí by mělo být na prvním místě šťastné dětství. Příliš mnoho vyšetření a jmenování lékaře zabere čas a během několika měsíců nedokáže problém genetické mutace prakticky vyřešit. Je proto vhodné, aby rodiče zacházeli se svým dítětem empaticky, ale aby ho v dospívání vychovali jako samostatného a sebevědomého. Otevřenost ohledně nemoci a rozhovory s postiženými o jejich zkušenostech, i když díky její vzácnosti je nepravděpodobné, že by to osobně ovlivnilo ostatní, bude společně pomáhat při řízení života.
Epidemiologie
Odhaduje se, že postihuje méně než jednoho z milionu lidí.[2] Dosud bylo popsáno pouze 50 až 100 případů.[2]
Dějiny
Onemocnění poprvé popsal v roce 1935 německý lékař Hans Wagner.[3]
Reference
- ^ VYHRAZENO, VLOŽTE NÁS 14-- VŠECHNA PRÁVA. „Orphanet: Hypotrichóza s juvenilní makulární degenerací“. www.orpha.net. Citováno 24. června 2019.
- ^ A b C „Hypotrichóza - juvenilní makuladegenerace: ORPHA1573“. Orphanet (v němčině). Citováno 2016-08-11.
- ^ Wagner, H. (1935). „Maculaaffektion, vergesellschaftet mit Haarabnormitat von Lanugotypus, beide vielleicht angeboren bei zwei Geschwistern“. Graefes Archiv Klinischer und Experimenteller Ophthalalmologie (v němčině). 134: 74–81. doi:10.1007 / BF01854763. S2CID 21073109.
Zdroje
- „Vzácný syndrom: Hypotrichóza s juvenilní makulární dystrofií (HJMD)“. Investigativní oftalmologie a vizuální věda. 55 (13): 6424. Duben 2014.
- Online Mendelian Inheritance in Man (OMIM): CADHERIN 3 - 114021
- Samuelov, L; Sprecher, E; Tsuruta, D; Bíró, T; Kloepper, J. E .; Paus, R (2012). „P-kadherin reguluje růst a cyklování lidských vlasů pomocí kanonické signalizace Wnt a transformace růstového faktoru-β2“. Journal of Investigative Dermatology. 132 (10): 2332–41. doi:10.1038 / jid.2012.171. PMID 22696062.
- Nagel-Wolfrum, K; Möller, F; Penner, já; Wolfrum, U (2014). „Translační čtení jako alternativní přístup k oční genové terapii retinálních dystrofií způsobené in-frame nesmyslnými mutacemi“. Vizuální neurověda. 31 (4–5): 309–16. doi:10.1017 / S0952523814000194. PMID 24912600. (Posouzení).
- Gregory-Evans, C. Y .; Wang, X; Wasan, K. M .; Zhao, J; Metcalfe, A. L .; Gregory-Evans, K (2014). „Postnatální manipulace s dávkováním Pax6 zvrací vrozené vady tkáně“. Journal of Clinical Investigation. 124 (1): 111–116. doi:10.1172 / JCI70462. PMC 3871240. PMID 24355924.
- Schwarz, N .; Carr, A.-J .; Lane, A .; Moeller, F .; Chen, L. L .; Aguila, M .; Nommiste, B .; Muthiah, M. N .; Kanuga, N .; Wolfrum, U .; Nagel-Wolfrum, K .; Da Cruz, L .; Coffey, P. J .; Cheetham, M. E.; Hardcastle, A. J. (2014). „Translační přečtení mutace RP2 Arg120 stop v pacientských buňkách retinálního pigmentového epitelu odvozeného od iPSC“. Lidská molekulární genetika. 24 (4): 972–86. doi:10,1093 / hmg / ddu509. PMC 4986549. PMID 25292197.
externí odkazy
| Klasifikace | |
|---|---|
| Externí zdroje |