Agregace proteinů - Protein aggregation

Agregace proteinů je biologický jev, ve kterém vnitřně neuspořádané proteiny nebo špatně složené bílkoviny agregovat (tj. hromadit se a shlukovat) buď intra- nebo extracelulárně.[1][2] Špatně složené proteinové agregáty často korelují s nemocemi. Ve skutečnosti byly proteinové agregáty zapojeny do celé řady nemocí známých jako amyloidózy, počítaje v to ALS, Alzheimerova choroba, Parkinsonova choroba a prion choroba.[3][4]
Po syntéze se proteiny obvykle skládají do konkrétní trojrozměrné konformace, která je termodynamicky nejpříznivější: jejich nativní stav.[5] Tento proces skládání je řízen hydrofobní účinek: tendence hydrofobních (vodou se obávajících) částí proteinu chránit se před hydrofilním (vodou milujícím) prostředím buňky zakopáním do vnitřku proteinu. Exteriér proteinu je tedy typicky hydrofilní, zatímco vnitřek je typicky hydrofobní.
Proteinové struktury jsou stabilizovány nekovalentní interakce a disulfidové vazby mezi dvěma cystein zbytky. Mezi nekovalentní interakce patří iontové a slabé interakce van der Waalsovy interakce. Iontové interakce se tvoří mezi aniontem a kationtem a tvoří solné můstky, které pomáhají stabilizovat protein. Van der Waalsovy interakce zahrnují nepolární interakce (tj. London disperzní síla ) a polární interakce (tj. Vodíkové vazby, dipól-dipólová vazba ). Ty hrají důležitou roli v sekundární struktuře proteinu, jako je tvorba alfa šroubovice nebo beta list a terciární struktura. Interakce mezi aminokyselinovými zbytky ve specifickém proteinu jsou velmi důležité v konečné struktuře tohoto proteinu.
Pokud dojde ke změnám v nekovalentních interakcích, jak se může stát při změně v aminokyselinové sekvenci, je protein náchylný k nesprávnému složení nebo rozvinutí. V těchto případech, pokud buňka nepomáhá proteinu při opětovném skládání nebo degradaci rozloženého proteinu, může se rozložený / nesprávně složený protein agregovat, ve kterém mohou exponované hydrofobní části proteinu interagovat s exponovanými hydrofobními náplastmi jiných proteinů .[6][7] Mohou se tvořit tři hlavní typy proteinových agregátů: amorfní agregáty, oligomery, a amyloid fibrily.[8]
Příčiny
K agregaci proteinů může docházet z různých příčin. Tyto příčiny lze rozdělit do čtyř tříd, které jsou podrobně popsány níže.
Mutace
Mutace které se vyskytují v sekvenci DNA, mohou nebo nemusí ovlivnit aminokyselinovou sekvenci proteinu. Když je ovlivněna sekvence, může odlišná aminokyselina měnit interakce mezi postranními řetězci, které ovlivňují skládání proteinu. To může vést k exponovaným hydrofobním oblastem proteinu, které agregují se stejným nesprávně složeným / rozloženým proteinem nebo jiným proteinem.
Kromě mutací v samotných postižených proteinech může být agregace proteinů způsobena také nepřímo prostřednictvím mutací v proteinech v regulačních drahách, jako je refoldingová dráha (molekulární chaperony ) nebo dráha ubikvitin-proteazom (ubikvitinové ligázy).[9] Chaperony pomoc s přeložením bílkovin zajištěním bezpečného prostředí pro složení bílkovin. Ubikvitinové ligázy cílí na proteiny pro degradaci prostřednictvím modifikace ubikvitinu.
Problémy se syntézou bílkovin
Agregace proteinů může být způsobena problémy, ke kterým dochází během transkripce nebo překlad. Během transkripce je DNA kopírována do mRNA, čímž se vytváří vlákno pre-mRNA, které prochází Zpracování RNA za vzniku mRNA.[10] Během překladu ribozomy a tRNA pomozte převést sekvenci mRNA na sekvenci aminokyselin.[10] Pokud během některého z kroků vzniknou problémy s vytvořením nesprávného řetězce mRNA a / nebo nesprávné aminokyselinové sekvence, může to způsobit nesprávné složení proteinu, což vede k agregaci proteinu.
Stresy prostředí
Stresy prostředí, jako jsou extrémní teploty a pH nebo oxidační stres může také vést k agregaci bílkovin.[11] Jednou z takových nemocí je kryoglobulinémie.
Extrémní teploty mohou oslabit a destabilizovat nekovalentní interakce mezi aminokyselinovými zbytky. pH mimo rozsah pH proteinu může změnit protonační stav aminokyselin, což může zvýšit nebo snížit nekovalentní interakce. To může také vést k méně stabilním interakcím a vést k rozvinutí bílkovin.
Oxidační stres může být způsoben radikály, jako jsou reaktivní formy kyslíku (ROS). Tyto nestabilní radikály mohou napadat aminokyselinové zbytky, což vede k oxidaci postranních řetězců (např. aromatický boční řetězy, methionin postranní řetězce) a / nebo štěpení polypeptidových vazeb.[12] To může ovlivnit nekovalentní interakce, které udržují protein pohromadě správně, což může způsobit destabilizaci proteinu a může způsobit odvíjení proteinu.[11]
Stárnutí
Buňky mají mechanismy, které mohou znovu skládat nebo degradovat proteinové agregáty. Jak však buňky stárnou, jsou tyto kontrolní mechanismy oslabeny a buňka je méně schopná rozložit agregáty.[11]
Hypotéza, že agregace proteinů je příčinným procesem stárnutí, je nyní testovatelná, protože existují některé modely opožděného stárnutí. Pokud byl vývoj proteinových agregátů procesem nezávislým na stárnutí, zpomalení stárnutí nebude mít v průběhu času žádný vliv na rychlost proteotoxicity. Pokud je však stárnutí spojeno s poklesem aktivity ochranných mechanismů proti proteotoxicitě, modely pomalého stárnutí by vykazovaly sníženou agregaci a proteotoxicitu. K řešení tohoto problému bylo provedeno několik testů toxicity C. elegansTyto studie ukázaly, že snížení aktivity signalizace inzulín / IGF (IIS), významná regulační cesta stárnutí chrání před agregací toxických proteinů spojenou s neurodegenerací. Platnost tohoto přístupu byla testována a potvrzena u savců, protože snížení aktivity signální dráhy IGF-1 chránilo Alzheimerovy modelové myši před behaviorálními a biochemickými poruchami spojenými s onemocněním.[13]
Souhrnná lokalizace
Několik studií ukázalo, že buněčné reakce na agregaci proteinů jsou dobře regulované a organizované. Agregáty proteinů se lokalizují do specifických oblastí v buňce a byl proveden výzkum těchto lokalizací v prokaryotech (E. coli) a eukaryotech (kvasinky, buňky savců).
Bakterie
Agregáty v bakteriích asymetricky končí na jednom z pólů buňky, „starším pólu“. Poté, co se buňka rozdělí, dceřiné buňky se starším pólem získají agregát bílkovin a rostou pomaleji než dceřiné buňky bez agregátu. To poskytuje mechanismus přirozené selekce pro redukci agregátů proteinů v bakteriální populaci.[14]
Droždí
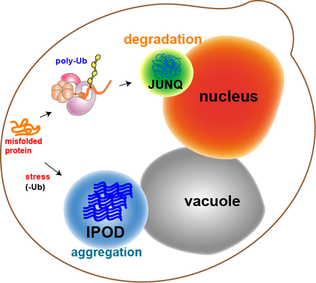
Většina proteinových agregátů v kvasinkových buňkách je znovu složena molekulárními chaperony. Některé agregáty, jako jsou oxidačně poškozené proteiny nebo proteiny označené pro degradaci, však nelze znovu složit. Spíše existují dva oddíly, ve kterých mohou skončit. Proteinové agregáty lze lokalizovat v oddělení kontroly kvality Juxtanuclear (JUNQ ), který je v blízkosti jaderné membrány nebo na ložisku nerozpustného proteinu (IPOD ), poblíž vakuoly v kvasinkových buňkách.[11] Agregáty proteinů se lokalizují na JUNQ, když jsou ubikvitinovány a cíleny na degradaci. Agregované a nerozpustné proteiny se lokalizují na IPOD jako trvalejší depozice. Existují důkazy, že proteiny zde mohou být odstraněny autofagií.[15] Tyto dvě cesty spolupracují v tom, že proteiny mají tendenci přicházet na IPOD, když je cesta proteazomu přepracována.[15]
Savčí buňky
V savčích buňkách se tyto proteinové agregáty nazývají „agresory“ a vytvářejí se, když je buňka nemocná. Je to proto, že agregáty mají tendenci se tvořit, když existují heterologní proteiny přítomné v buňce, které mohou vzniknout při mutaci buňky. E3 ubikvitinová ligáza je schopna rozpoznat špatně složené proteiny a ubikvitovat je. HDAC6 se pak může vázat na ubikvitin a motorický protein dynein přivést označené agregáty do organizačního centra mikrotubulů (MTOC ). Tam se sbalí do koule, která obklopuje MTOC. Přinášejí chaperony a proteazomy a aktivují autofagii.[16]
Odstranění
V buňce existují dva hlavní systémy kontroly kvality bílkovin, které jsou odpovědné za eliminaci proteinových agregátů. Špatně poskládané proteiny mohou být znovu složeny bi-chaperonovým systémem nebo degradovány ubikvitinovým proteazomovým systémem nebo autofagií.[17]
Přeložení
Bi-chaperonový systém využívá Hsp70 (DnaK-DnaJ-GrpE v E. coli a Ssa1-Ydj1 / Sis1-Sse1 / Fe1 v kvasinkách) a Hsp100 (ClpB v E. coli a Hsp104 v kvasinkách) chaperony pro disagregaci a opětovné složení proteinů .[18]
Hsp70 interaguje s agregáty proteinů a rekrutuje Hsp100. Hsp70 stabilizuje aktivovaný Hsp100. Proteiny Hsp100 mají smyčky aromatických pórů, které se používají pro vláknovou aktivitu k rozmotání jednotlivých polypeptidů. Tato vláknová aktivita může být zahájena na N-konci, C-konci nebo ve středu polypeptidu. Polypeptid se translokuje prostřednictvím Hsp100 v řadě kroků, přičemž v každém kroku využívá ATP.[18] Polypeptid se rozloží a poté se nechá znovu složit sám nebo pomocí proteinů tepelného šoku.[19]
Degradace
Špatně poskládané proteiny lze eliminovat systémem ubikvitin-proteazom (UPS ). Skládá se z dráhy E1-E2-E3, která ubikvinuje proteiny a označuje je pro degradaci. U eukaryot se proteiny degradují proteasomem 26S. V savčích buňkách se E3 ligáza, karboxyterminální protein interagující s Hsp70 (CHIP), zaměřuje na proteiny vázané na Hsp70. V kvasinkách mají E3 ligázy Doa10 a Hrd1 podobné funkce endoplazmatické retikulum bílkoviny.[20]

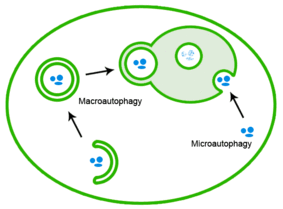
Špatně poskládané proteiny lze také eliminovat autofagií, při které jsou proteinové agregáty dodávány do lysozomu.[20]
Toxicita
Ačkoli se předpokládá, že samotné agregáty zralých proteinů jsou toxické, nedávné důkazy naznačují, že ve skutečnosti jsou toxické agregáty nezralých proteinů.[21][22] Hydrofobní náplasti těchto agregátů mohou interagovat s jinými složkami buňky a poškodit je. Předpokládá se, že toxicita proteinových agregátů souvisí s mechanismy sekvestrace buněčných složek, tvorbou reaktivních forem kyslíku a vazbou na specifické receptory v membráně nebo narušením membrán.[23] Kvantitativní test byl použit k určení, že za membránovou permeaci jsou odpovědné druhy s vyšší molekulovou hmotností.[24] Je známo, že proteinové agregáty in vitro mohou destabilizovat umělé fosfolipidové dvojvrstvy, což vede k permeabilizaci membrány.
Viz také
Reference
- ^ Aguzzi, A .; O'Connor, T. (březen 2010). "Choroby agregace proteinů: patogenita a terapeutické perspektivy". Recenze přírody Objev drog. 9 (3): 237–48. doi:10.1038 / nrd3050. PMID 20190788. S2CID 5756683.
- ^ Stefani, M .; Dobson, CM. (Listopad 2003). „Agregace proteinů a toxicita agregátů: nový pohled na skládání proteinů, špatně skládané nemoci a biologickou evoluci“. J Mol Med (Berl). 81 (11): 678–99. doi:10.1007 / s00109-003-0464-5. PMID 12942175. S2CID 23544974.
- ^ De Felice, FG .; Vieira, MN .; Meirelles, MN .; Morozova-Roche, LA .; Dobson, CM .; Ferreira, ST. (Červenec 2004). "Tvorba agregátů amyloidu z lidského lysozymu a jeho variant souvisejících s onemocněním pomocí hydrostatického tlaku". FASEB J.. 18 (10): 1099–101. doi:10.1096 / fj.03-1072fje. PMID 15155566. S2CID 13647147.
- ^ Tanzi, RE .; Bertram, L. (únor 2005). „Dvacet let hypotézy amyloidů Alzheimerovy choroby: genetická perspektiva“. Buňka. 120 (4): 545–55. doi:10.1016 / j.cell.2005.02.008. PMID 15734686. S2CID 206559875.
- ^ Brüning, Ansgar; Jückstock, Julia (01.01.2015). „Špatně poskládané bílkoviny: od malých darebáků po malé pomocníky v boji proti rakovině“. Hranice v onkologii. 5: 47. doi:10,3389 / fonc.2015.00047. PMC 4338749. PMID 25759792.
- ^ Gething, MJ .; Sambrook, J. (leden 1992). "Složení bílkovin v buňce". Příroda. 355 (6355): 33–45. Bibcode:1992 Natur.355 ... 33G. doi:10.1038 / 355033a0. PMID 1731198. S2CID 4330003.
- ^ Roberts, CJ. (Prosinec 2007). "Kinetika agregace nepůvodních proteinů". Biotechnol Bioeng. 98 (5): 927–38. doi:10,1002 / bit. 21627. PMID 17705294. S2CID 21787377.
- ^ Cox, David L .; Nelson, Michael M. (2013). Lehningerovy principy biochemie. New York: W.H. Freemane. str. 143. ISBN 978-1-4292-3414-6.
- ^ Berke, Sarah J. Shoesmith; Paulson, Henry L (06.06.2003). "Agregace proteinů a dráha ubikvitin proteazomu: získání UPPer ruky na neurodegeneraci". Aktuální názor na genetiku a vývoj. 13 (3): 253–261. doi:10.1016 / S0959-437X (03) 00053-4. PMID 12787787.
- ^ A b Weaver, Robert F. (2012). Molekulární biologie. New York: McGraw-Hill. str. 122–156, 523–600. ISBN 978-0-07-352532-7.
- ^ A b C d Tyedmers, Jens; Mogk, Axel; Bukau, Bernd (listopad 2010). "Buněčné strategie pro řízení agregace proteinů". Nature Reviews Molecular Cell Biology. 11 (11): 777–788. doi:10.1038 / nrm2993. PMID 20944667. S2CID 22449895.
- ^ Stadtman, E. R .; Levine, R. L. (2003-07-29). „Oxidace volných aminokyselin a aminokyselinových zbytků v proteinech zprostředkovaná volnými radikály“. Aminokyseliny. 25 (3–4): 207–218. doi:10.1007 / s00726-003-0011-2. ISSN 0939-4451. PMID 14661084. S2CID 26844881.
- ^ Morley JF, Brignull HR, Weyers JJ, Morimoto RI (2002). „Prahová hodnota pro agregaci proteinů s polyglutaminovou expanzí a buněčnou toxicitu je dynamická a ovlivněna stárnutím v Caenorhabditiselegans“. PNAS. 99 (16): 10417–10422. Bibcode:2002PNAS ... 9910417M. doi:10.1073 / pnas.152161099. PMC 124929. PMID 12122205.
- ^ Bednarska, Natalia G .; Schymkowitz, Joost; Rousseau, Frederic; Van Eldere, Johan (01.01.2013). „Agregace proteinů v bakteriích: tenká hranice mezi funkčností a toxicitou“. Mikrobiologie. 159 (9): 1795–1806. doi:10.1099 / mic.0.069575-0. PMID 23894132.
- ^ A b Takalo, Mari; Salminen, Antero; Soininen, Hilkka; Hiltunen, Mikko; Haapasalo, Annakaisa (8. 3. 2013). "Mechanismy agregace a degradace proteinů u neurodegenerativních onemocnění". American Journal of Neurodegenerative Disease. 2 (1): 1–14. ISSN 2165-591X. PMC 3601466. PMID 23516262.
- ^ Garcia-Mata, Rafael; Gao, Ya-Sheng; Sztul, Elizabeth (01.06.2002). "Potýká se s vyřazováním odpadků: zhoršující se agresivita". Provoz. 3 (6): 388–396. doi:10.1034 / j.1600-0854.2002.30602.x. ISSN 1600-0854. PMID 12010457. S2CID 305786.
- ^ Gregersen, Niels; Bolund, Lars; Bross, Peter (01.10.2005). "Špatné skládání, agregace a degradace proteinů u nemoci". Molekulární biotechnologie. 31 (2): 141–150. doi:10,1385 / MB: 31: 2: 141. ISSN 1073-6085. PMID 16170215. S2CID 36403914.
- ^ A b Mogk, Axel; Kummer, Eva; Bukau, Bernd (01.01.2015). „Spolupráce chaperonových strojů Hsp70 a Hsp100 při disagregaci proteinů“. Frontiers in Molecular Biosciences. 2: 22. doi:10.3389 / fmolb.2015.00022. ISSN 2296-889X. PMC 4436881. PMID 26042222.
- ^ Liberek, Krzysztof; Lewandowska, Agnieszka; Ziętkiewicz, Szymon (23. ledna 2008). „Chaperony kontrolující disagregaci proteinů“. Časopis EMBO. 27 (2): 328–335. doi:10.1038 / sj.emboj.7601970. ISSN 0261-4189. PMC 2234349. PMID 18216875.
- ^ A b Chen, Bryan; Retzlaff, Marco; Roos, Thomas; Frydman, Judith (01.08.2011). „Buněčné strategie kontroly kvality bílkovin“. Perspektivy Cold Spring Harbor v biologii. 3 (8): a004374. doi:10.1101 / cshperspect.a004374. ISSN 1943-0264. PMC 3140689. PMID 21746797.
- ^ Zhu, YJ .; Lin, H .; Lal, R. (červen 2000). „Čerstvý a nefibrilární amyloidový beta protein (1–40) indukuje rychlou buněčnou degeneraci ve starých lidských fibroblastech: důkaz buněčné toxicity zprostředkované AbetaP kanálem“. FASEB J.. 14 (9): 1244–54. doi:10.1096 / fasebj.14.9.1244. PMID 10834946. S2CID 42263619.
- ^ Nilsberth, C .; Westlind-Danielsson, A .; Eckman, CB .; Condron, MM .; Axelman, K .; Forsell, C .; Stenh, C .; Luthman, J .; Teplow, DB .; et al. (Září 2001). „„ Arktická “mutace APP (E693G) způsobuje Alzheimerovu chorobu zvýšenou tvorbou protofibril Abeta.“. Nat Neurosci. 4 (9): 887–93. doi:10.1038 / nn0901-887. PMID 11528419. S2CID 13516479.
- ^ Soto C (2003). „Rozvíjení role nesprávného skládání bílkovin u neurodegenerativních onemocnění“. Nat. Rev. Neurosci. 4 (1): 49–60. doi:10.1038 / nrn1007. PMID 12511861. S2CID 205499427.
- ^ Flagmeier P, De S, Wirthensohn DC, Lee SF, Vincke C, Muyldermans S, Knowles TPJ, Gandhi S, Dobson CM, Klenerman D (2017). „Ultrasenzitivní měření přítoku Ca2 + do lipidových váčků indukovaných agregáty proteinů“. Angew. Chem. Int. Vyd. Angl. 56 (27): 7750–7754. doi:10.1002 / anie.201700966. PMC 5615231. PMID 28474754.