Alfa-aminoadipová semialdehyd syntáza - Alpha-aminoadipic semialdehyde synthase






Alfa-aminoadipová semialdehyd syntáza je enzym kódováno AASS gen u lidí a podílí se na jejich hlavní činnosti lysin degradační cesta. Je podobný samostatným enzymům kódovaným pro geny LYS1 a LYS9 v kvasinkách a souvisí s, i když není podobnou strukturou, bifunkční enzym nachází se v rostlinách.[5][6] U lidí jsou mutace v genu AASS a odpovídající alfa-aminoadipická enzym semialdehyd syntáza spojeny s familiární hyperlysinemie.[5][7][8] Tato podmínka se dědí v autosomální recesivní vzor a není považován za zvlášť negativní stav, takže je vzácný choroba.[9]
Funkce
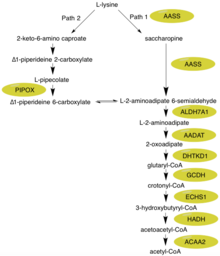
Alfa-aminoadipická semialdehyd syntáza protein katalyzuje první dva kroky v savčím L-lysin degradace prostřednictvím sacharopin cesta v rámci mitochondrie, o kterém se předpokládá, že je hlavní metabolickou cestou pro degradaci lysinu v horní části eukaryoty.[11][12] Specifická subpathway, na kterou se tento enzym zaměřuje, je syntéza glutaryl-CoA z L-lysinu.[9] Glutaryl-CoA může působit jako meziprodukt v rozšířenější cestě konverze / degradace z L-lysinu na acetyl-CoA.
Mezi viditelnými složkami degradace L-lysinu cestou sacharopinu jsou středně používaná reakce / produkt glutamát a případný záchyt uhlíku acetyl-CoA. Glutamát je důležitá sloučenina v těle, která působí jako neurotransmiter spojený s učením a Huntingtonovou chorobou.[13][14] Acetyl-CoA má pravděpodobně ještě vyšší úroveň důležitosti, působí jako jedna z integrálních složek cyklu kyselina citronová / Kreb, s primární funkcí dodávat acetylovou skupinu, která má být oxidována pro výrobu energie.[15] Funkce alfa-aminoadipické semialdehyd syntázy je tedy vázána na hladiny dvou integrálních sloučenin v těle.
Mechanismus

Nejprve N-terminál část tohoto enzymu, která obsahuje lysin -ketoglutarát reduktáza (LOR / LKR) aktivita (ES: 1.5.1.8) kondenzuje lysin a 2-oxoglutarát na molekulu nazývanou sacharopin (reakce 1 na obrázku vpravo).[7][11] Poté C-terminál část tohoto enzymu, která obsahuje sacharopin dehydrogenáza (SHD) aktivita (EC: 1.5.1.9), katalyzuje oxidaci sacharopinu za vzniku alfa-aminoadipová semialdehyd a glutamát (reakce 2 na obrázku vpravo).[7][11] Poznámka: Tyto reakce jsou opakem příslušných kroků v drahách biosyntézy lysinu přítomných v droždí a houby.[16][17][18]
Tyto reakce lze vizualizovat také ve formě reakční rovnice:
N (6) - (L-1,3-dikarboxypropyl) -L-lysin + NADP+ + H2O = L-lysin + 2-oxoglutarát + NADPH následovaný
N (6) - (L-1,3-dikarboxypropyl) -L-lysin + NAD+ + H2O = L-glutamát + (S) -2-amino-6-oxohexanoát + NADH.[9]
Struktura
Přirozený lidský enzym je bifunkční, podobně jako LKR / SHD nalezený v rostlinách, a proto se předpokládá, že má podobnou strukturu.[16] Bifunkčnost tohoto enzymu pochází ze skutečnosti, že obsahuje dvě odlišná aktivní místa, jedno na jeho C-konci a jedno na jeho N-konci.[7] C-koncová část alfa-aminoadipické semialdehyd syntázy obsahuje aktivitu SHD a N-koncová část obsahuje LKR.[19] K dnešnímu dni nebyla stanovena struktura alfa-aminoadipické semialdehyd syntázy.[20] Enzym nemá spojovací oblast přítomnou v rostlinách mezi jeho C a N-konci, takže teorie naznačují, že skutečná struktura obsahuje oblast aktivity LKR vázanou na oblast aktivity SHD, jako je tomu v Magnaporthe grisea.[19]

Relevance nemoci
Alfa-aminoadipová semialdehyd syntáza je kódována genem AASS a mutace v tomto genu vedou k hyperlysinemii.[5][7] To je charakterizováno zhoršeným rozkladem lysinu, který vede ke zvýšeným hladinám lysinu v krvi a moči. Zdá se, že tyto zvýšené hladiny lysinu nemají žádné negativní účinky na tělo.[8] Jiné názvy této podmínky zahrnují:[8]
- onemocnění z nedostatku alfa-aminoadipického semialdehydu
- familiární hyperlysinemie
- onemocnění způsobené nedostatkem lysin alfa-ketoglutarát reduktázy
- onemocnění způsobené nedostatkem sacharopin dehydrogenázy
- sacharopinurie
Hyperlysinemie je charakterizována zvýšenými plazmatickými hladinami lysinu, které přesahují 600 μmol / l a mohou dosáhnout až 2 000 μmol / l.[21][22] Zdá se, že tyto zvýšené hladiny lysinu nemají žádné negativní účinky na tělo.[8] Hlavním důvodem je to, že může probíhat několik alternativních biochemických reakcí. Nejprve může být lysin použit místo ornithinu v močovinovém cyklu, což vede k produkci homoargininu.[23] Navíc, i když většina savců používá pro většinu degradace lysinu dráhu sacharopinu (Cesta 1), mozek má alternativní cestu (Cesta 2), která prochází meziproduktem kyseliny L-pipekolové - obojí je vidět na obrázku.[23] Je důležité si uvědomit, že cesta 1 se odehrává v mitochondriích, zatímco cesta 2 se odehrává v peroxisomu.[12] Při pohledu na další klíčové enzymy v cestě degradace L-lysinu je ALDH7A1 deficitní u dětí se záchvaty závislými na pyridoxinu.[24] GCDH má nedostatek glutarové acidurie typu 1.[25] Meziprodukt 2-oxoadipát je metabolizován 2-oxoadipát dehydrogenázou, což připomíná 2-oxoglutarát dehydrogenázu z komplexu enzymu kyselina citronová / Krebův cyklus.[10]
Dosud byly popsány dva typy familiární hyperlysinémie: typ I je spojen s kombinovaným nedostatkem dvou enzymových aktivit, LOR a SDH, zatímco u familiární hyperlysinemie typu II je narušena pouze aktivita sacharopin dehydrogenázy.[26][27] Hyperlysinemie typu II se také označuje jako sacharopinurie.[10]
Dalším stavem, který je prokázán v souvislosti s hyperlysinemií, je nedostatek dienoyl-CoA reduktázy, ačkoli se jedná o relativně nedávný objev a není k dispozici mnoho publikací, které by to podporovaly.[28]
Reference
- ^ A b C GRCh38: Vydání souboru 89: ENSG00000008311 - Ensembl, Květen 2017
- ^ A b C GRCm38: Vydání souboru 89: ENSMUSG00000029695 - Ensembl, Květen 2017
- ^ „Human PubMed Reference:“. Národní centrum pro biotechnologické informace, Americká národní lékařská knihovna.
- ^ „Myš PubMed Reference:“. Národní centrum pro biotechnologické informace, Americká národní lékařská knihovna.
- ^ A b C Sacksteder KA, Biery BJ, Morrell JC, Goodman BK, Geisbrecht BV, Cox RP, Gould SJ, Geraghty MT (červen 2000). „Identifikace genu pro alfa-aminoadipickou semialdehyd syntázu, který je vadný u familiární hyperlysinemie“. American Journal of Human Genetics. 66 (6): 1736–43. doi:10.1086/302919. PMC 1378037. PMID 10775527.
- ^ Zhu X, Tang G, Galili G (prosinec 2002). „Aktivita bifunkčního enzymu lysin-ketoglutarátreduktáza / sacharopindehydrogenáza Arabidopsis lysinového katabolismu je regulována funkční interakcí mezi jeho dvěma doménami enzymů.“. The Journal of Biological Chemistry. 277 (51): 49655–61. doi:10,1074 / jbc.M205466200. PMID 12393892.
- ^ A b C d E „Entrez Gene: AASS aminoadipate-semialdehyde synthase“.
- ^ A b C d „hyperlysinemie“. Genetická domácí reference. Citováno 2017-03-04.
- ^ A b C "Alpha-aminoadipic semialdehyde syntáza, mitochondriální". UniProt. Citováno 2017-03-04.
- ^ A b C Houten SM, Te Brinke H, Denis S, Ruiter JP, Knegt AC, de Klerk JB, Augoustides-Savvopoulou P, Häberle J, Baumgartner MR, Coşkun T, Zschocke J, Sass JO, Poll-The BT, Wanders RJ, Duran M (Duben 2013). "Genetický základ hyperlysinémie". Orphanet Journal of Rare Diseases. 8: 57. doi:10.1186/1750-1172-8-57. PMC 3626681. PMID 23570448.
- ^ A b C Papes F, Kemper EL, Cord-Neto G, Langone F, Arruda P (prosinec 1999). „Odbourávání lysinu cestou sacharopinu u savců: účast bifunkčních i monofunkčních enzymů degradujících lysin u myší“. The Biochemical Journal. 344 (2): 555–63. doi:10.1042/0264-6021:3440555. PMC 1220675. PMID 10567240.
- ^ A b Danhauser K, Sauer SW, Haack TB, Wieland T, Staufner C, Graf E, Zschocke J, Strom TM, Traub T, Okun JG, Meitinger T, Hoffmann GF, Prokisch H, Kölker S (prosinec 2012). „Mutace DHTKD1 způsobují 2-aminoadipovou a 2-oxoadipovou acidurii“. American Journal of Human Genetics. 91 (6): 1082–7. doi:10.1016 / j.ajhg.2012.10.006. PMC 3516599. PMID 23141293.
- ^ Meldrum BS (duben 2000). „Glutamát jako neurotransmiter v mozku: přehled fyziologie a patologie“. The Journal of Nutrition. 130 (4S Suppl): 1007S – 15S. doi:10.1093 / jn / 130.4.1007s. PMID 10736372. Archivovány od originál dne 2017-05-16. Citováno 2017-03-05.
- ^ „O toxicitě glutamátu“. Hunting Disease Outreach for Education at Stanford (HOPES). Huntington's Disease Society of America. Citováno 2017-03-05.
- ^ Ophardt CE (2003). „Sloučenina acetyl CoA Crossroads“. Virtuální ChemBook. Elmhurst College.
- ^ A b Markovitz PJ, Chuang DT, Cox RP (říjen 1984). "Familiární hyperlysinemie. Čištění a charakterizace bifunkční aminoadipové semialdehyd syntázy s aktivitou lysin-ketoglutarát reduktázy a sacharopin dehydrogenázy". The Journal of Biological Chemistry. 259 (19): 11643–6. PMID 6434529.
- ^ Jones EE, Broquist HP (červen 1965). „Saccharopine, meziprodukt dráhy aminoadipové kyseliny biosyntézy lysinu. II. Studie na Saccharomyces cereviseae.“ The Journal of Biological Chemistry. 240: 2531–6. PMID 14304864.
- ^ Trupin JS, Broquist HP (červen 1965). „Saccharopin, meziprodukt dráhy aminokyseliny biosyntézy lysinu v kyselině aminoadipové. I. studie na neurospora crassa“. The Journal of Biological Chemistry. 240: 2524–30. PMID 14304863.
- ^ A b Johansson E, Steffens JJ, Lindqvist Y, Schneider G (říjen 2000). „Krystalová struktura sacharopin reduktázy z Magnaporthe grisea, enzymu alfa-aminoadipátové dráhy biosyntézy lysinu“. Struktura. 8 (10): 1037–47. doi:10.1016 / s0969-2126 (00) 00512-8. PMID 11080625.
- ^ "AASS - aminoadipát-semialdehyd syntáza". RCSB Proteinová datová banka - prostřednictvím PDB.
- ^ Hoffmann GF, Kolker S (2012). "Poruchy mozkových organických kyselin a jiné poruchy katabolismu lysinu". V Saudubray JM, van den Berghe G, Walter JH (eds.). Diagnostika a léčba vrozených metabolických chorob (5. vydání). Berlín: Springer. 333–346. ISBN 978-3-642-15720-2.
- ^ Saudubray JM, Rabier D (červen 2007). "Biomarkery identifikované u vrozených chyb lysinu, argininu a ornithinu". The Journal of Nutrition. 137 (6 Suppl 2): 1669S – 1672S. doi:10.1093 / jn / 137.6.1669S. PMID 17513445.
- ^ A b vd Heiden C, Brink M, de Bree PK, v Sprang FJ, Wadman SK, de Pater JM, van Biervliet JP (1978). "Familiární hyperlysinémie způsobená nedostatkem L-lysin alfa-ketoglutarát reduktázy: výsledky pokusu o léčbu". Journal of Inherited Metabolic Disease. 1 (3): 89–94. doi:10.1007 / bf01805679. PMID 116084. S2CID 35326745.
- ^ Mills PB, Struys E, Jakobs C, Plecko B, Baxter P, Baumgartner M, Willemsen MA, Omran H, Tacke U, Uhlenberg B, Weschke B, Clayton PT (březen 2006). „Mutace v antikvitinu u jedinců se záchvaty závislými na pyridoxinu“. Přírodní medicína. 12 (3): 307–9. doi:10,1038 / nm1366. PMID 16491085. S2CID 27940375.
- ^ Goodman SI, Kratz LE, DiGiulio KA, Biery BJ, Goodman KE, Isaya G, Frerman FE (září 1995). „Klonování cDNA glutaryl-CoA dehydrogenázy a exprese divokého typu a mutantních enzymů v Escherichia coli“. Lidská molekulární genetika. 4 (9): 1493–8. doi:10,1093 / hmg / 4.9.1493. PMID 8541831.
- ^ Dancis J, Hutzler J, Cox RP (květen 1979). „Familiární hyperlysinemie: studie enzymů, diagnostické metody, komentáře k terminologii“. American Journal of Human Genetics. 31 (3): 290–9. PMC 1685795. PMID 463877.
- ^ Cederbaum SD, Shaw KN, Dancis J, Hutzler J, Blaskovics JC (srpen 1979). „Hyperlysinemie se sacharopinurií způsobená kombinovanými nedostatky lysin-ketoglutarát reduktázy a sacharopin dehydrogenázy, které se projevují jako cystinurie“. The Journal of Pediatrics. 95 (2): 234–8. doi:10.1016 / s0022-3476 (79) 80657-5. PMID 571908.
- ^ Houten SM, Denis S, Te Brinke H, Jongejan A, van Kampen AH, Bradley EJ, Baas F, Hennekam RC, Millington DS, Young SP, Frazier DM, Gucsavas-Calikoglu M, Wanders RJ (září 2014). „Mitochondriální nedostatek NADP (H) způsobený mutací v NADK2 způsobuje nedostatek dienoyl-CoA reduktázy s hyperlysinemií“. Lidská molekulární genetika. 23 (18): 5009–16. doi:10,1093 / hmg / ddu218. PMID 24847004.
Další čtení
- Maruyama K, Sugano S (leden 1994). „Oligo-capping: jednoduchá metoda k nahrazení struktury cap eukaryotických mRNA oligoribonukleotidy“. Gen. 138 (1–2): 171–4. doi:10.1016/0378-1119(94)90802-8. PMID 8125298.
externí odkazy
- AASS umístění lidského genu v UCSC Genome Browser.
- AASS podrobnosti o lidském genu v UCSC Genome Browser.