Živá polymerace - Living polymerization
v polymerní chemie, živá polymerace je forma řetězová polymerace kde schopnost růstu polymerní řetězec na vypovědět byla odstraněna.[1][2] Toho lze dosáhnout různými způsoby. Ukončení řetězce a reakce řetězového přenosu chybí a míra zahájení řetězce je také mnohem větší než míra šíření řetězce. Výsledkem je, že polymerní řetězce rostou více konstantní hodnotit než je vidět v tradičním řetězci polymerizace a jejich délky zůstávají velmi podobné (tj. mají velmi nízkou index polydisperzity ). Živá polymerace je populární metoda syntézy blokové kopolymery protože polymer může být syntetizován ve fázích, přičemž každá fáze obsahuje jinou monomer. Další výhody jsou předem stanoveny molární hmotnost a kontrolu nad koncové skupiny.
Živá polymerace je žádoucí, protože nabízí přesnost a kontrolu v makromolekulární syntéze. To je důležité, protože mnoho nových / užitečných vlastností polymerů vyplývá z jejich mikrostruktury a molekulové hmotnosti. Od té doby molekulární váha a disperzita jsou méně kontrolované v neživých polymeracích, je tato metoda vhodnější pro návrh materiálů[3][4]
V mnoha případech jsou živé polymerační reakce zaměňovány nebo považovány za synonyma kontrolovaných polymerací. I když jsou tyto polymerační reakce velmi podobné, existuje výrazný rozdíl v definicích těchto dvou reakcí. Zatímco živé polymerizace jsou definovány jako polymerační reakce, při kterých je eliminace ukončení nebo přenosu řetězce, řízené polymerační reakce jsou reakce, při nichž je ukončení potlačeno, ale ne eliminováno, zavedením klidového stavu polymeru.[3][4] Toto rozlišení je však v literatuře stále předmětem debaty.
Hlavní živé polymerační techniky jsou:
- Živá aniontová polymerace
- Živá kationtová polymerace
- Živobytí polymerace s otevřením kruhu
- Živá polymerace volných radikálů
- Živé polykondenzace růstu řetězce
Dějiny
Živou polymeraci demonstroval Michael Szwarc v roce 1956 aniontovou polymerací styren s alkalický kov / naftalen systém v tetrahydrofuran (THF). Szwarc to ukázal elektronový přenos došlo od radikální anion z naftalen na styren. Počáteční radikální anion styrenu se převádí na a dianion (nebo ekvivalentně disodio-) druhy, které rychle přidávaly styren za vzniku „dvousložkového živého polymeru“. Důležitým aspektem jeho práce byl Szwarc aprotické rozpouštědlo tetrahydrofuran, který se rozpouští, ale je jinak nereaktivní vůči organokovovým meziproduktům. Po počátečním přidání monomeru do iniciátorového systému se viskozita by se zvýšil (kvůli zvýšenému růstu polymerního řetězce), ale nakonec přestal po vyčerpání koncentrace monomeru. Zjistil však, že přidání více monomer způsobil zvýšení viskozity, což naznačuje růst polymerního řetězce, a tak dospěl k závěru, že polymerní řetězce nebyly nikdy ukončeny.[6] To byl hlavní krok v polymerní chemii, protože kontrola nad tím, kdy byl polymer zastaven nebo ukončen, obecně nebyl kontrolovaným krokem. S tímto objevem se seznam potenciálních aplikací dramaticky rozšířil.[7]
Dnes se živé polymerace široce používají při výrobě mnoha druhů polymerů nebo plastů. Tento přístup nabízí kontrolu nad chemickým složením polymeru, a tím i nad strukturálními a elektronickými vlastnostmi materiálu. Tato úroveň kontroly zřídka existuje v neživých polymeračních reakcích.[4][8]
Rychlá iniciace: nízká polydisperzita


Jednou z klíčových charakteristik živé polymerace je to, že reakce ukončení a přenosu řetězce jsou v podstatě eliminovány ze čtyř elementárních reakcí řetězová polymerace pouze odchod zahájení a (řetězové) propagační reakce.
Klíčovou charakteristikou živé polymerace je to, že rychlost iniciace (což znamená, že spící chemické látky generují aktivní látky šířící řetězec) je mnohem rychlejší než rychlost šíření řetězce. Všechny řetězce tedy rostou stejnou rychlostí (rychlostí šíření).
Vysoká rychlost iniciace (spolu s absencí ukončení) vede k nízkému (nebo úzkému) indexu polydisperzity (PDI), což je známkou široké distribuce polymerních řetězců (Živé polymery ) Prodloužená životnost množícího se řetězce, která umožňuje provádět na živém řetězci tvorbu kopolymeru polymeru a funkčnost koncové skupiny. Tyto faktory také umožňují předvídatelné molekulové hmotnosti, vyjádřené jako číselná průměrná molekulová hmotnost (Mn). Pro ideální životní systém je předpoklad účinnosti generování aktivních druhů 100%, kdy každý iniciátor generuje pouze jeden aktivní druh Kinetická délka řetězu (průměrný počet monomerů, se kterými aktivní druh reaguje během své životnosti) v daném čase lze odhadnout pomocí znalosti koncentrace zbývajícího monomeru. Číselná průměrná molekulová hmotnost, Mn, se zvyšuje lineárně s procentem konverze během živé polymerace
![v={frac {[M]_{0}-[M]}{[I]_{0}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/db304320640f163330c3130db03ff9691e1a8ffa)
Techniky
Živá aniontová polymerace
Již v roce 1936 Karl Ziegler navrhl, že aniontová polymerace styrenu a butadienu postupným přidáváním monomeru k alkyl lithiovému iniciátoru proběhla bez přenosu řetězce nebo ukončení. O dvacet let později Szwarc demonstroval živou polymeraci prostřednictvím aniontová polymerace z styren v THF použitím naftalenid sodný jako iniciátor.[9][6][10]
Naftalenový anion iniciuje polymeraci redukcí styrenu na jeho radikální anion, který dimerizuje na dilithiodifenylbutan, který pak iniciuje polymeraci. Tyto experimenty se opíraly o Szwarcovu schopnost řídit hladinu nečistot, které by zničily vysoce reaktivní organokovové meziprodukty.
Živá polymerace α-olefinů
α-olefiny mohou být polymerovány aniontovou koordinační polymerací, při které je kovové centrum katalyzátoru považováno za protikation pro aniontový konec alkylového řetězce (prostřednictvím koordinace M-R). Iniciátory Ziegler-Natta byly vyvinuty v polovině 50. let a jsou heterogenními iniciátory používanými při polymeraci alfa-olefinů. Nejen, že tito iniciátoři jako první dosáhli poly (1-alkenů) s relativně vysokou molekulovou hmotností (v současnosti nejrozšířenější termoplast na světě PE (Polyethylen ) a PP (Polypropylen )[11] ale iniciátoři byli také schopni stereoselktivních polymerací, které se připisují chirální Krystalická struktura heterogenního iniciátoru.[4] Vzhledem k důležitosti tohoto objevu byly Ziegler a Natta představeny 1963 Nobelova cena za chemii. Ačkoli aktivní druhy vytvořené z iniciátoru Ziegler-Natta mají obecně dlouhou životnost (v rozsahu hodin nebo déle), životnost rozmnožovacích řetězců se zkracuje kvůli několika drahám přenosu řetězce (Vylučování beta-hydridu a převod na spoluiniciátora) a ve výsledku se nepovažují za žijící.[4]
Metalocenové iniciátory jsou považovány za typ iniciátorů Ziegler-Natta díky použití dvousložkového systému sestávajícího z přechodový kov a kovový koiniciátor skupiny I-III (například methylalumoxan (MAO) nebo jiné alkylaluminiové sloučeniny). The metalocen iniciátory tvoří homogenní jediné místo katalyzátory které byly původně vyvinuty ke studiu dopadu struktury katalyzátoru na výslednou strukturu / vlastnosti polymerů; což bylo obtížné pro heterogenní iniciátory Ziegler-Natta s více místy.[11] Díky diskrétnímu jednomu místu na metalocenovém katalyzátoru byli vědci schopni vyladit a spojit, jak struktura pomocného ligandu (ty, které se přímo nepodílejí na chemických transformacích) a symetrie kolem centra chirálního kovu ovlivňují mikrostrukturu polymeru.[12] Avšak v důsledku reakcí na rozrušení řetězce (hlavně eliminace beta-hydridu) je známo velmi málo polymerací na bázi metalocenu.[4]
Vyladěním sterického objemu a elektronických vlastností pomocných ligandů a jejich substituenty třída iniciátorů známá jako chelátovat iniciátory (nebo post-metalocenové iniciátory) byly úspěšně použity stereospecifické živé polymerace alfa-olefinů. Chelátové iniciátory mají vysoký potenciál pro živé polymerace, protože pomocné ligandy mohou být navrženy tak, aby odradily nebo inhibovaly dráhy ukončení řetězce. Chelátové iniciátory lze dále rozložit na základě pomocných ligandů; ansa-cyklopentyadienyl-amido iniciátory, cheláty alfa-diiminu a cheláty fenoxyiminu.[4]
- Ansa-cyklopentadienyl-amido (CpA) iniciátory

Iniciátoři CpA jeden mají cyklopentadienyl substituent a jeden nebo více dusíkatých substituentů koordinovaných ke středu kovu (obecně Zr nebo Ti) (Odian). Dimetyl (pentamethylcyklopentyl) zirkoniumacetamidinát na obrázku___ byl použit pro stereospecifickou živou polymeraci 1-hexenu při -10 ° C. Výsledný poly (1-hexen) byl izotaktický (stereohemie je stejná mezi sousedními opakujícími se jednotkami) potvrzeno 13C-NMR. Několik pokusů prokázalo kontrolovatelnost a předvídatelnost (od katalyzátoru po monomer poměr) Mn s nízkým Đ. Polymerace byla dále potvrzena jako živá postupným přidáváním 2 částí monomeru, druhá část byla přidána poté, co již byla první část polymerována, a monitorování D a Mn řetězu. Výsledné polymerní řetězce vyhovovaly předpokládané Mn (s celkovou koncentrací monomeru = část 1 +2) a vykazovaly nízkou hodnotu D[13] což naznačuje, že řetězce byly stále aktivní nebo živé, protože byla přidána druhá část monomeru (5).
- α-diimin chelátové iniciátory
Iniciátory chelátů a-diiminů se vyznačují tím, že mají a diimin chelatující struktura pomocného ligandu a která je obecně koordinována do centra kovu s pozdním přechodem (tj. Ni a Pd).

Brookhart a kol. provedli rozsáhlou práci s touto třídou katalyzátorů a uvedli živou polymeraci pro α-olefiny[14] a demonstrovali živé alternativní kopolymery oxidu uhelnatého a-olefinu.[15]
Živá kationtová polymerace
Monomery pro živou kationtovou polymeraci jsou elektrony bohaté alkeny, jako jsou vinylethery, isobutylen, styren a N-vinylkarbazol. Iniciátory jsou binární systémy skládající se z elektrofilu a Lewisovy kyseliny. Tato metoda byla vyvinuta kolem roku 1980 za přispění Higashimury, Sawamota a Kennedyho. Vytvoření stabilní karbokationtu po delší dobu je obvykle obtížné kvůli možnosti, že se kation rozloží p-protony připojenými k jinému monomeru v hlavním řetězci nebo ve volném monomeru. Proto se používá jiný přístup[3][4][16]

V tomto příkladu je karbokace generována přidáním Lewisovy kyseliny (koiniciátor spolu s halogenem „X“ již na polymeru - viz obrázek), který nakonec generuje karbokation ve slabé rovnováze. Tato rovnováha silně zvýhodňuje spící stav, takže zbývá málo času na trvalé zhášení nebo ukončení jinými cestami. Kromě toho lze také přidat slabý nukleofil (Nu :), aby se ještě více snížila koncentrace aktivních druhů, čímž polymer zůstane „živý“.[3][4][16] Je však důležité si to uvědomit podle definice polymery popsané v tomto příkladu nejsou technicky živé kvůli zavedení neaktivního stavu, protože ukončení bylo pouze sníženo, nikoli odstraněno (i když toto téma je stále předmětem debaty). Fungují však podobně a používají se v podobných aplikacích jako aplikace skutečných živých polymerací.
Žijící polymerace polymerace otevírající kruh
Při správných reakčních podmínkách polymerace s otevřením kruhu (ROMP) lze učinit živým. První takové systémy popsal Robert H. Grubbs v roce 1986 na základě norbornene a Tebbeho činidlo a v roce 1978 Grubbs společně s Richard R. Schrock popisující živou polymeraci s a wolfram karbenový komplex.[17]
Obecně reakce ROMP zahrnují konverzi cyklického olefinu s významným kmenem v kruhu (> 5 kcal / mol), jako je cyklobuten, norbornen, cyklopenten atd., Na polymer, který také obsahuje dvojné vazby. Důležitá věc, kterou je třeba si povšimnout o polymeracích při otevírání kruhu, je, že dvojná vazba je obvykle udržována v páteři, což jí umožňuje považovat ji za „živou“ za správných podmínek.[18]
Aby byla reakce ROMP považována za „živou“, je třeba splnit několik pokynů:[18]
- Rychlá a úplná iniciace monomeru. To znamená, že rychlost, jakou iniciační činidlo aktivuje monomer pro polymeraci, musí nastat velmi rychle.
- Kolik monomerů tvoří každý polymer (stupeň polymerace) musí souviset lineárně s množstvím monomeru, se kterým jste začali.
- The disperzita polymeru musí být <1,5. Jinými slovy, distribuce toho, jak dlouho jsou vaše polymerní řetězce ve vaší reakci, musí být velmi nízká.
S ohledem na tyto pokyny vám umožňuje vytvořit polymer, který je dobře kontrolován jak obsahem (jaký monomer používáte), tak vlastnostmi polymeru (což lze do značné míry přičíst délce polymerního řetězce). Je důležité si uvědomit, že živé polymerace otevírající kruh mohou být aniontové nebo kationtový.

Protože u živých polymerů byla odstraněna jejich terminační schopnost, znamená to, že jakmile bude váš monomer spotřebován, přidání dalšího monomeru způsobí pokračování růstu polymerních řetězců, dokud nebude spotřebován veškerý další monomer. To bude pokračovat, dokud se kovový katalyzátor na konci řetězce záměrně neodstraní přidáním zhášecího činidla. Ve výsledku může potenciálně umožnit vytvoření a blok nebo gradientový kopolymer poměrně snadno a přesně. To může vést k vysoké schopnosti vyladit vlastnosti polymeru na požadovanou aplikaci (elektrické / iontové vedení atd.)[4][18]
„Živá“ polymerace volných radikálů
Počínaje sedmdesátými léty bylo objeveno několik nových metod, které umožňovaly vývoj živé polymerace pomocí volné radikály chemie. Tyto techniky zahrnovaly katalytický přenos řetězce polymerace, polymerace zprostředkovaná iniferterem, stabilní polymerace zprostředkovaná volnými radikály (SFRP), atomová radikálová polymerace (ATRP), reverzibilní přenos řetězce a fragmentace (VOR ) polymerace a polymerace s přenosem jódu.
Při "živé" radikálové polymeraci (nebo řízené radikálové polymeraci (CRP)) jsou dráhy rozbíjení řetězců ve srovnání s konvenční radikální polymerací (RP) vážně sníženy a CRP může vykazovat vlastnosti živé polymerace. Protože však ukončení řetězce chybí, ale pouze je minimalizováno, CRP technicky nesplňuje požadavky kladené IUPAC na živou polymeraci (viz úvod k definici IUPAC). O tomto čísle se diskutuje a názory různých vědců lze najít ve zvláštním čísle časopisu Journal of Polymer Science Žijící nebo ovládaní?. Tato otázka dosud nebyla v literatuře vyřešena, proto je často označována jako „živá“ polymerace, kvazi-živá polymerace, pseudoživá a další pojmy pro označení této problematiky.
V CRP se používají dvě obecné strategie k potlačení řetězových reakcí a k podpoře rychlé iniciace ve vztahu k šíření. Obě strategie jsou založeny na rozvoji dynamické rovnováhy mezi aktivním množitelským radikálem a spícím druhem.[19]
První strategie zahrnuje reverzibilní zachycovací mechanismus, ve kterém se šířící radikál podrobí aktivaci / deaktivaci (tj. Radikální polymerace přenosu atomů ) proces s druhem X. Druh X je perzistentní radikál nebo druh, který může generovat stabilní radikál, který se nemůže ukončit sám sebou nebo se množit, ale pouze reverzibilně „ukončit“ množícím se radikálem (z množícího se polymerního řetězce) P *. P * je radikální druh, který se může množit (kp) a nevratně ukončit (kt) s jiným P *. X je normálně nitroxid (tj. TEMPO použito v Radikální polymerace zprostředkovaná nitroxidem ) nebo organokovový druh. Spící druh (P.n-X) lze aktivovat k regeneraci aktivních množitelských druhů (P *) spontánně, tepelně, za použití katalyzátoru a opticky.[19][20]
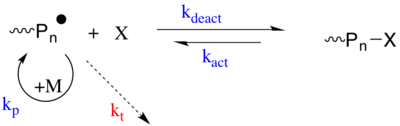
Druhá strategie je založena na degenerativním přenosu (DT) množícího se radikálu mezi přenosovým činidlem, které působí jako spící druh (tj. Reverzibilní adice - fragmentace polymerace řetězovým přenosem ). CRP založené na DT sledují konvenční kinetiku radikálové polymerace, tj. Pomalou iniciaci a rychlé ukončení, ale přenosové činidlo (Pm-X nebo Pn-X) je přítomno v mnohem vyšší koncentraci ve srovnání s radikálovým iniciátorem. Množící se radikální druhy procházejí tepelně neutrální výměnou se spícím přenosovým činidlem chemickým přenosem atomů, skupin nebo adičních fragmentů.[19]

Živé polykondenzace růstu řetězce
Polykondenzační polymerace pro růst řetězce byly původně vyvinuty za předpokladu, že změna substitučních účinků polymeru ve srovnání s monomerem způsobí, že koncová skupina polymerů bude reaktivnější, což se označuje jako „reaktivní intermediární polykondenzace“. Zásadním výsledkem je, že monomery přednostně reagují s koncovými skupinami aktivovaného polymeru před reakcemi s jinými monomery. Tato upřednostňovaná reaktivita je zásadním rozdílem při kategorizaci polymeračního mechanismu na rozdíl od řetězového růstu krokový růst ve kterém monomerní a polymerní koncová skupina řetězce mají stejnou reaktivitu (reaktivita je nekontrolovaná). Bylo použito několik strategií k minimalizaci reakcí monomer-monomer (nebo samokondenzace) a tímto mechanismem u polymerů s malou molekulovou hmotností bylo dosaženo polymerací s nízkým D a kontrolovatelným Mn.[21] U vysokomolekulárních polymerních řetězců (tj. Malého poměru iniciátoru k monomeru) však Mn není u některých monomerů snadno regulovatelný, protože častěji dochází ke kondenzaci mezi monomery kvůli nízké koncentraci druhů.[21]
Polykondenzace s přenosem katalyzátoru
Polykondenzace přenosu katalyzátoru (CTP) je mechanismus polykondenzace růstu řetězce, při kterém monomery přímo nereagují navzájem a místo toho bude monomer reagovat pouze s koncovou skupinou polymeru prostřednictvím mechanismu zprostředkovaného katalyzátorem.[21] Obecný postup spočívá v tom, že katalyzátor aktivuje koncovou skupinu polymeru, po které následuje reakce koncové skupiny s druhým příchozím monomerem. Katalyzátor se poté přenese do podlouhlého řetězce při aktivaci koncové skupiny (jak je znázorněno níže).[22]

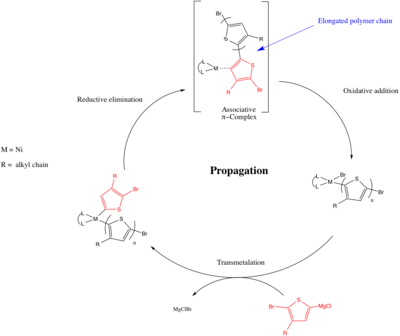
Polykondenzace přenosu katalyzátoru umožňuje živou polymeraci polymerů konjugovaných s π a byla objevena Tsutomu Yokozawou v roce 2004[22] a Richard McCullough.[23] V CTP je krok šíření založen na organických křížových vazebných reakcích (tj. Spojka Kumada, Sonogashira spojka, Spojka Negishi ) vrchní forma vazby uhlík-uhlík mezi difunkčními monomery. Když Yokozawa a McCullough nezávisle objevili polymeraci pomocí kovového katalyzátoru, který spojil a Grignardovo činidlo s organohalogenidem vytvářejícím novou vazbu uhlík-uhlík. Níže uvedený mechanismus ukazuje tvorbu poly (3-alkylthiofenu) za použití Ni iniciátoru (L.n může být 1,3-Bis (difenylfosfino) propan (dppp) ) a je podobný konvenčnímu mechanismu pro Spojka Kumada zahrnující oxidační přísada, a transmetalace a a redukční eliminace krok. Existuje však klíčový rozdíl, po redukční eliminaci v CTP se vytvoří asociativní komplex (který byl podpořen intra- / intermolekulárními experimenty s oxidační adiční konkurencí[8]) a k následnému oxidačnímu přidání dochází mezi kovovým středem a přidruženým řetězcem (intramolekulární dráha). Zatímco při kopulační reakci nově vytvořená alkyl / arylová sloučenina difunduje a dochází k následnému oxidačnímu přidání mezi příchozí vazbou Ar – Br a středem kovu. Asociativní komplex je nezbytný pro to, aby polymerace probíhala živým způsobem, protože umožňuje, aby kov podstoupil výhodný intramolekulární oxidační přídavek a zůstal s jediným množícím se řetězcem (v souladu s mechanismem růstu řetězce), na rozdíl od mezimolekulárního oxidačního přídavku s další monomery přítomné v roztoku (v souladu s mechanismem postupného růstu, neživého).[24][25] Rozsah monomeru CTP se od jeho objevování zvyšuje a zahrnuje poly (fenylen) s, poly (fluor), s, poly (selenofen) a poly (pyrrol).[24][25]
Polymerace přenosu živých skupin
Skupinová polymerace má také vlastnosti živé polymerace.[26] Aplikuje se na alkylované methakrylát monomery a iniciátorem je a silylketenacetal. Nový monomer se přidává k iniciátoru a aktivnímu rostoucímu řetězci v a Michaelova reakce. Při každém přidání monomerní skupiny se trimethylsilylová skupina přenese na konec řetězce. Aktivní konec řetězu není iontový jako při aniontové nebo kationtové polymeraci, ale je kovalentní. Reakce může být katalyzována bifluoridy a bioxyanionty, jako jsou tris (dialkylamino) sulfonium bifluorid nebo tetrabutylamoniumbibenzoát. Metodu objevil v roce 1983 O.W. Webster[27] a jméno nejprve navrhl Barry Trost.
Aplikace
Živé polymerace se používají při komerční syntéze mnoha polymerů.
Syntéza a aplikace kopolymeru
Kopolymery jsou polymery sestávající z několika různých druhů monomerů a mohou být uspořádány v různých pořadích, z nichž tři jsou vidět na obrázku níže.

I když existují další (střídavé kopolymery, roubované kopolymery a stereoblokové kopolymery), tyto tři jsou ve vědecké literatuře běžnější.[3] Kromě toho mohou blokové kopolymery existovat v mnoha typech, včetně tribloku (A-B-A), střídavého bloku (A-B-A-B-A-B) atd.
Z těchto tří typů se blokové a gradientové kopolymery běžně syntetizují prostřednictvím živých polymerací, a to díky snadné kontrole, kterou poskytuje živá polymerace. Kopolymery jsou velmi žádané kvůli zvýšené flexibilitě vlastností, které polymer může mít ve srovnání s jejich homopolymerními protějšky. Použité syntetické techniky sahají od ROMP po generické aniontové nebo kationtové živé polymerace.[3][4]
Kopolymery mohou mít díky své jedinečné laditelnosti vlastností širokou škálu aplikací. Jedním z příkladů (mnoha) je nanoměřítko litografie pomocí blokových kopolymerů. Často používaným je blokový kopolymer vyrobený z polystyrenu a poly (methylmethakrylátu) (zkráceně PS-b-PMMA). Tento kopolymer může při správných tepelných a procesních podmínkách vytvářet válce o průměru několika desítek nanometrů v průměru PMMA obklopené maticí PS. Tyto válce lze poté odleptat při vysokém vystavení UV záření a kyselině octové, čímž se získá porézní matrice PS.[28][29][30]
Jedinečnou vlastností tohoto materiálu je, že velikost pórů (nebo velikost válců PMMA) lze snadno vyladit poměrem PS k PMMA při syntéze kopolymeru. To lze snadno vyladit díky snadné kontrole dané živými polymeračními reakcemi, což činí tuto techniku vysoce žádanou pro různé nanoscale vzorování různých materiálů pro aplikace na katalýzu, elektroniku atd.
Reference
- ^ Halasa, A. F. (1981). "Nedávné pokroky v aniontové polymeraci". Chemie a technologie kaučuku. 54 (3): 627–640. doi:10.5254/1.3535823.
- ^ Moad, Graeme and Solomon, David H. (2006) Chemie radikální polymerace. 2. vyd. Elsevier. ISBN 0-08-044286-2
- ^ A b C d E F Cowie, J.M.G. (2007). Chemie polymerů a fyzika moderních materiálů (3. vydání / J.M.G. Cowie a Valeria Arrighi ed.). Boca Raton: Taylor & Francis. ISBN 9780849398131.
- ^ A b C d E F G h i j k l Odian, George (2004). Principy polymerace (4. vyd.). Hoboken, NJ: Wiley-Interscience. ISBN 978-0471274001.
- ^ Jenkins, A. D .; Kratochvíl, P .; Stepto, R. F. T .; Suter, U. W. (1996). „Glosář základních pojmů v polymerní vědě (doporučení IUPAC 1996)“. Čistá a aplikovaná chemie. 68 (12): 2287–2311. doi:10.1351 / pac199668122287.
- ^ A b Szwarc, M .; Levy, M .; Milkovich, R. (1956). „Polymerizace iniciovaná elektronovým přenosem na monomer. Nová metoda tvorby blokových polymerů1“. Journal of the American Chemical Society. 78 (11): 2656–2657. doi:10.1021 / ja01592a101.
- ^ M. Szwarc (1956). ""Živé „polymery“. Příroda. 178 (4543): 1168. doi:10.1038 / 1781168a0.
- ^ A b McNeil, Anne; Bryan, Zachary (2013). „Důkazy o preferenčním intramolekulárním oxidačním přídavku v Ni-katalyzovaných křížových vazebných reakcích a jejich dopadu na polymerace růstu řetězce“. Chem. Sci. 4 (4): 1620–1624. doi:10.1039 / C3SC00090G.
- ^ Szwarc, M. (1956). "'Živé „polymery“. Příroda. 178 (4543): 1168–1169. Bibcode:1956 Natur.178.1168S. doi:10.1038 / 1781168a0.
- ^ Tatemoto, Masayoshi a Nakagawa, Tsuneo „Segmentované polymery obsahující fluor a jod a jejich výroba“ US patent 4 158 678 . Datum priority 30. června 1976.
- ^ A b Craver, C .; Carraher, C. (2000). Aplikovaná polymerní věda: 21. století. Elsevier. str. 1022–1023.
- ^ Coates, Geoffrey W. (duben 2000). „Přesná kontrola stereochemie polyolefinů pomocí kovových katalyzátorů na jednom místě“. Chemické recenze. 100 (4): 1223–1252. doi:10.1021 / cr990286u. PMID 11749265.
- ^ Jayaratne, K .; Sita, L (2000). „Stereospecifická žijící Ziegler-Natta polymerace 1-hexenu“. J. Am. Chem. Soc. 122 (5): 958–959. doi:10.1021 / ja993808w.
- ^ Killian, C. M .; Tempel, D. J .; Johnson, L. K.; Brookhart, M. (1996). „Živá polymerizace α-olefinů pomocí NiII-α-diiminových katalyzátorů. Syntéza nových blokových polymerů na bázi α-olefinů“. Journal of the American Chemical Society. 118 (46): 11664–11665. doi:10.1021 / ja962516h.
- ^ Brookhart, M .; Rix, F. C .; Desimone, J. M .; Barborak, J. C. (1992). „Palladium (II) katalyzátory pro živou střídavou kopolymeraci olefinů a oxidu uhelnatého“. Journal of the American Chemical Society. 114 (14): 5894–5895. doi:10.1021 / ja00040a082.
- ^ A b Goethals, E; Duprez, F (2007). "Carbocationic polymerisation". Pokrok ve vědě o polymerech. 32 (2): 220–246. doi:10.1016 / j.progpolymsci.2007.01.001.
- ^ Schrock, R.R .; Feldman, J .; Cannizzo, L. F .; Grubbs, R. H. (1987). "Polymerace norbornenu otevírající kruh živým komplexem alkyliden wolframu". Makromolekuly. 20 (5): 1169–1172. Bibcode:1987MaMol..20.1169S. doi:10.1021 / ma00171a053.
- ^ A b C Bielawski, Christopher W .; Grubbs, Robert H. (2007). "Polymerizace živého prstence otevírající metatézu". Pokrok ve vědě o polymerech. 32 (1): 1–29. doi:10.1016 / j.progpolymsci.2006.08.006.
- ^ A b C Braunecker, Wade A .; Matyjaszewski, Krzysztof (2007). "Řízená / živá radikálová polymerace: Vlastnosti, vývoj a perspektivy". Pokrok ve vědě o polymerech. 32 (1): 93–146. doi:10.1016 / j.progpolymsci.2006.11.002.
- ^ Matyjaszewski. "Vlastnosti řízené" živé "polymerizace". Archivovány od originál dne 14. března 2014.
- ^ A b C Yokozawa, T .; Yokoyama, A. (2007). „Chain-growth polycondensation: the living polymerization process in polycondensation“. Pokrok ve vědě o polymerech. 32: 147–172. doi:10.1016 / j.progpolymsci.2006.08.001.
- ^ A b Miyakoshi, Ryo; Yokoyama, Akihiro; Yokozawa, Tsutomu (2005). „Polykondenzace s přenosem katalyzátoru. Mechanismus polymerace Ni-katalyzovaného řetězového růstu vedoucí k dobře definovanému poly (3-hexylthiofenu)“. Journal of the American Chemical Society. 127 (49): 17542–17547. doi:10.1021 / ja0556880. PMID 16332106.
- ^ Iovu, Mihaela Corina; Sheina, Elena E .; Gil, Roberto R .; McCullough, Richard D. (říjen 2005). „Experimentální důkazy pro kvazi -„ živou “povahu metody Grignardovy metathézy pro syntézu regioregulárních poly (3-alkylthiofenů)“. Makromolekuly. 38 (21): 8649–8656. Bibcode:2005MaMol..38,8649I. CiteSeerX 10.1.1.206.3875. doi:10.1021 / ma051122k.
- ^ A b Kiriy, Anton; Senkovskij, Volodymyr; Sommer, Michael (4. října 2011). „Polykondenzace katalyzátoru a přenosu Kumada: mechanismus, příležitosti a výzvy“. Makromolekulární rychlá komunikace. 32 (19): 1503–1517. doi:10.1002 / březen.201100316. PMID 21800394.
- ^ A b Bryan, Zachary J .; McNeil, Anne J. (12. listopadu 2013). „Syntéza konjugovaných polymerů pomocí polykondenzace s přenosem katalyzátoru (CTP): mechanismus, rozsah a aplikace“. Makromolekuly. 46 (21): 8395–8405. Bibcode:2013MaMol..46,8395B. doi:10.1021 / ma401314x.
- ^ Davis, Fred J. (2004) Chemie polymerů: praktický přístup. Oxford University Press. ISBN 978-0-19-850309-5 .
- ^ Webster, O. W .; Hertler, W. R .; Sogah, D. Y .; Farnham, W. B .; RajanBabu, T. V. (1983). „Polymerace skupinovým přenosem. 1. Nový koncept adiční polymerace s iniciátory organokřemíku“. J. Am. Chem. Soc. 105 (17): 5706–5708. doi:10.1021 / ja00355a039.
- ^ In, Insik; La, Young-Hye; Park, Sang-Min; Nealey, Paul F .; Gopalan, Padma (srpen 2006). „Náhodné kopolymerové štětce s bočním řetězcem jako neutrální povrchy pro řízení orientace mikrodomén blokového kopolymeru v tenkých vrstvách“. Langmuir. 22 (18): 7855–7860. doi:10,1021 / la060748g. PMID 16922574.
- ^ Liu, Yuanjun; Gong, Yanchun; On, Longbin; Xie, Bo; Chen, Xi; Han, Min; Wang, Guanghou (2010). „Tvorba periodických polí nanorování na samo-sestaveném PS-b-PMMA filmu pod rychlým žíháním rozpouštědlem“. Nanoměřítko. 2 (10): 2065–8. Bibcode:2010Nanos ... 2.2065L. doi:10.1039 / c0nr00207k. PMID 20820641.
- ^ Edwards, E. W .; Montague, M. F .; Solak, H. H .; Hawker, C. J .; Nealey, P. F. (4. srpna 2004). „Přesná kontrola nad molekulárními dimenzemi domén blokových kopolymerů s využitím mezifázové energie chemicky nanopatternovaných substrátů“. Pokročilé materiály. 16 (15): 1315–1319. doi:10.1002 / adma.200400763.