Biopython - Biopython
| Původní autoři | Chapman B, Chang J.[1] |
|---|---|
| První vydání | 2000[1] |
| Stabilní uvolnění | 1.74[2] / 16. července 2019 |
| Úložiště | https://github.com/biopython/biopython |
| Napsáno | Krajta a C |
| Plošina | Cross-platform |
| Typ | Bioinformatika |
| Licence | Licence Biopython |
| webová stránka | biopython |
The Biopython projekt je open-source sbírka nekomerčních Krajta nástroje pro výpočetní biologie a bioinformatika, vytvořené mezinárodní asociací vývojářů.[1][3][4] Obsahuje třídy, které mají být reprezentovány biologické sekvence a anotace sekvence a je schopen číst a zapisovat do různých formátů souborů. Umožňuje také programové prostředky přístupu online databáze biologických informací, jako například u NCBI. Samostatné moduly rozšiřují možnosti Biopythonu na zarovnání sekvence, proteinová struktura, populační genetika, fylogenetika, sekvenční motivy, a strojové učení. Biopython je jedním z řady projektů Bio * určených k redukci duplikace kódu v výpočetní biologie.[5]
Dějiny
Vývoj Biopython začal v roce 1999 a poprvé byl vydán v červenci 2000.[6] Byl vyvinut v podobném časovém rámci as podobnými cíli jako jiné projekty, které přidaly bioinformatické schopnosti do jejich příslušných programovacích jazyků, včetně BioPerl, BioRuby a BioJava. Mezi první vývojáře projektu patřili Jeff Chang, Andrew Dalke a Brad Chapman, přestože doposud přispělo více než 100 lidí.[7] V roce 2007 podobný Krajta projekt, jmenovitě PyCogent, byl založen.[8]
Počáteční rozsah Biopythonu zahrnoval přístup, indexování a zpracování souborů biologických sekvencí. I když je to stále hlavní zaměření, v následujících letech přidané moduly rozšířily jeho funkčnost tak, aby pokrývaly další oblasti biologie (viz Klíčové vlastnosti a příklady ).
Od verze 1.62 podporuje Biopython běh na Pythonu 3 i Pythonu 2.[9]
Design
Kdykoli je to možné, Biopython dodržuje konvence používané programovacím jazykem Python, aby usnadnil uživatelům obeznámeným s Pythonem. Například, Sekv a SeqRecord s objekty lze manipulovat pomocí krájení, podobným způsobem jako řetězce a seznamy Pythonu. Je také navržen tak, aby byl funkčně podobný jiným projektům Bio *, jako je BioPerl.[6]
Biopython je schopen číst a zapisovat nejběžnější formáty souborů pro každou ze svých funkčních oblastí a jeho licence je tolerantní a kompatibilní s většinou ostatních softwarových licencí, které umožňují použití Biopythonu v různých softwarových projektech.[4]
Klíčové vlastnosti a příklady
Sekvence
Základní koncepcí v Biopythonu je biologická sekvence, kterou reprezentuje Sekv třída.[10] Biopython Sekv objekt je v mnoha ohledech podobný řetězci Pythonu: podporuje notaci řezu Python, lze jej zřetězit s jinými sekvencemi a je neměnný. Kromě toho zahrnuje metody specifické pro sekvenci a specifikuje konkrétní použitou biologickou abecedu.
>>> # Tento skript vytváří sekvenci DNA a provádí některé typické manipulace>>> z Bio. Sek import Sekv>>> z Bio. Abeceda import IUPAC>>> dna_sequence = Sekv('AGGCTTCTCGTA', IUPAC.jednoznačná_dna)>>> dna_sequenceSeq ('AGGCTTCTCGTA', IUPACUnambiguousDNA ())>>> dna_sequence[2:7]Seq ('GCTTC', IUPACUnambiguousDNA ())>>> dna_sequence.reverse_complement()Seq ('TACGAGAAGCCT', IUPACUnambiguousDNA ())>>> rna_sequence = dna_sequence.přepsat()>>> rna_sequenceSeq ('AGGCUUCUCGUA', IUPACUnambiguousRNA ())>>> rna_sequence.přeložit()Seq ('RLLV', IUPACProtein ())Sekvenční anotace
The SeqRecord třída popisuje sekvence spolu s informacemi, jako je název, popis a funkce ve formě SeqFeature předměty. Každý SeqFeature objekt určuje typ prvku a jeho umístění. Typy prvků mohou být ‚gen ',‚ CDS' (kódovací sekvence), ‚repeat_region ',‚ mobile_element' nebo jiné a pozice prvků v sekvenci může být přesná nebo přibližná.
>>> # Tento skript načte anotovanou sekvenci ze souboru a zobrazí část jeho obsahu.>>> z Bio import Sekv>>> seq_record = Sekv.číst('pTC2.gb', 'genbank')>>> seq_record.název„NC_019375“>>> seq_record.popis"Providencia stuartii plasmid pTC2, úplná sekvence.">>> seq_record.funkce[14]SeqFeature (FeatureLocation (ExactPosition (4516), ExactPosition (5336), řetězec = 1), type = 'mobile_element')>>> seq_record.náslSeq ('GGATTGAATATAACCGACGTGACTGTTACATTTAGGTGGCTAAACCCGTCAAGC ... GCC', IUPACAmbiguousDNA ())Vstup a výstup
Biopython umí číst a zapisovat do řady běžných formátů sekvencí, včetně FASTA, FASTQ, GenBank, Clustal, PHYLIP a NEXUS. Při čtení souborů se popisné informace v souboru používají k naplnění členů tříd Biopython, například SeqRecord. To umožňuje převést záznamy jednoho formátu souboru do jiných.
Velmi velké soubory sekvence mohou překročit paměťové zdroje počítače, takže Biopython poskytuje různé možnosti přístupu k záznamům ve velkých souborech. Mohou být načteny úplně do paměti v datových strukturách Pythonu, jako jsou seznamy nebo slovníky, poskytující rychlý přístup za cenu využití paměti. Soubory lze případně číst z disku podle potřeby, s pomalejším výkonem, ale nižšími nároky na paměť.
>>> # Tento skript načte soubor obsahující více sekvencí a každou z nich uloží v jiném formátu.>>> z Bio import Sekv>>> genomy = Sekv.analyzovat('salmonella.gb', 'genbank')>>> pro genom v genomy:... Sekv.psát si(genom, genom.id + '.fasta', 'fasta')Přístup k online databázím
Prostřednictvím modulu Bio.Entrez mohou uživatelé Biopythonu stahovat biologická data z databází NCBI. Každá z funkcí poskytovaných Entrez vyhledávací modul je k dispozici prostřednictvím funkcí v tomto modulu, včetně vyhledávání a stahování záznamů.
>>> # Tento skript stáhne genomy z databáze NCBI Nucleotide a uloží je do souboru FASTA.>>> z Bio import Entrez>>> z Bio import Sekv>>> výstupní soubor = otevřeno('all_records.fasta', "w")>>> Entrez.e-mailem = '[email protected]'>>> records_to_download = [„FO834906.1“, „FO203501.1“]>>> pro record_id v records_to_download:... Rukojeť = Entrez.efetch(db=‚nukleotid ', id=record_id, rettype='gb')... seqRecord = Sekv.číst(Rukojeť, formát='gb')... Rukojeť.zavřít()... výstupní soubor.psát si(seqRecord.formát('fasta'))Fylogeneze

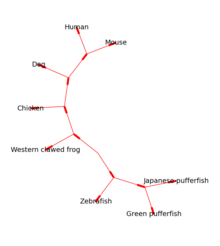
Modul Bio.Phylo poskytuje nástroje pro práci a vizualizaci fylogenetické stromy. Pro čtení a zápis jsou podporovány různé formáty souborů Newick, NEXUS a phyloXML. Běžné manipulace a procházení stromů jsou podporovány prostřednictvím Strom a Clade předměty. Mezi příklady patří převod a řazení souborů stromu, extrahování podmnožin ze stromu, změna kořene stromu a analýza funkcí větve, jako je délka nebo skóre.[12]
Zakořeněné stromy lze vtáhnout dovnitř ASCII nebo pomocí matplotlib (viz obrázek 1) a Graphviz Knihovna může být použita k vytvoření nezakořeněných rozvržení (viz obrázek 2).
Genomové diagramy
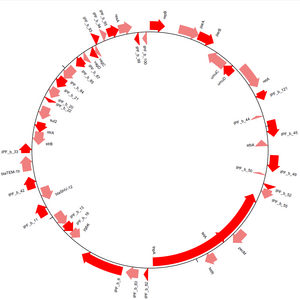
Modul GenomeDiagram poskytuje metody vizualizace sekvencí v Biopythonu.[14] Sekvence lze kreslit v lineární nebo kruhové formě (viz obrázek 3) a je podporována řada výstupních formátů, včetně PDF a PNG. Schémata se vytvářejí vytvářením stop a následným přidáním funkcí sekvencí k těmto stopám. Opakováním funkcí sekvence a použitím jejich atributů k rozhodování, zda a jak jsou přidány do stop diagramu, je možné vykonat velkou kontrolu nad vzhledem konečného diagramu. Křížové odkazy lze kreslit mezi různými stopami, což umožňuje jedné porovnávat více sekvencí v jednom diagramu.
Makromolekulární struktura
Modul Bio.PDB může načítat molekulární struktury z PDB a mmCIF soubory a byl přidán do Biopythonu v roce 2003.[15] The Struktura objekt je pro tento modul ústředním bodem a organizuje makromolekulární strukturu hierarchicky: Struktura objekty obsahují Modelka objekty, které obsahují Řetěz objekty, které obsahují Zbytek objekty, které obsahují Atom předměty. Neuspořádané zbytky a atomy mají své vlastní třídy, DisorderedResidue a DisorderedAtom, které popisují jejich nejisté polohy.
Pomocí Bio.PDB lze procházet jednotlivé složky souboru makromolekulární struktury, například zkoumat každý atom v proteinu. Lze provádět běžné analýzy, jako je měření vzdáleností nebo úhlů, porovnání zbytků a výpočet hloubky zbytků.
Populační genetika
Modul Bio.PopGen přidává podporu pro Biopython pro Genepop, softwarový balíček pro statistickou analýzu populační genetiky.[16] To umožňuje provádět analýzy Hardy – Weinbergova rovnováha, vazebná nerovnováha a další rysy populace frekvence alel.
Tento modul může také provádět populační genetické simulace pomocí koalescenční teorie s programem fastsimcoal2.[17]
Obálky pro nástroje příkazového řádku
Mnoho modulů Biopython obsahuje obálky příkazového řádku pro běžně používané nástroje, což umožňuje použití těchto nástrojů přímo z Biopythonu. Mezi tyto obaly patří VÝBUCH, Clustal, PhyML, VYTEPAT a SAMtools. Uživatelé mohou podtřídu třídy obecných obalů přidat podporu pro jakýkoli jiný nástroj příkazového řádku.
Viz také
Reference
- ^ A b C Chapman, Brad; Chang, Jeff (srpen 2000). "Biopython: Pythonové nástroje pro výpočetní biologii". Newsletter ACM SIGBIO. 20 (2): 15–19. doi:10.1145/360262.360268. S2CID 9417766.
- ^ „Vydán Biopython 1.74“. Otevřená nadace pro bioinformatiku. 16. července 2019.
- ^ Kohout, Peter JA; Antao, Tiago; Chang, Jeffery T; Chapman, Brad A; Cox, Cymon J; Dalke, Andrew; Friedberg, Iddo; Hamelryck, Thomas; Kauff, Frank; Wilczynski, Bartek; de Hoon, Michiel JL (20. března 2009). „Biopython: volně dostupné nástroje Pythonu pro výpočetní molekulární biologii a bioinformatiku“. Bioinformatika. 25 (11): 1422–3. doi:10.1093 / bioinformatika / btp163. PMC 2682512. PMID 19304878.
- ^ A b Další informace najdete na webových stránkách Biopython práce popisující Biopython a seznam více než sto publikace využívající / citující Biopython.
- ^ Mangalam, Harry (září 2002). „Sada nástrojů Bio * - stručný přehled“. Briefings in Bioinformatics. 3 (3): 296–302. doi:10.1093 / bib / 3.3.296. PMID 12230038.
- ^ A b Chapman, Brad (11. března 2004), Projekt Biopython: Filozofie, funkčnost a fakta (PDF), vyvoláno 11. září 2014
- ^ Seznam přispěvatelů Biopython, archivovány z originál dne 11. září 2014, vyvoláno 11. září 2014
- ^ Knight, R; Maxwell, P; Birmingham, A; Carnes, J; Caporaso, J. G .; Easton, B. C .; Eaton, M; Hamady, M; Lindsay, H; Liu, Z; Lozupone, C; McDonald, D; Robeson, M; Sammut, R; Smit, S; Wakefield, M. J .; Widmann, J; Wikman, S; Wilson, S; Ying, H; Huttley, G. A. (2007). „Py Přesvědčivý: Sada nástrojů pro dávání smyslu ze sekvence ". Genome Biology. 8 (8): R171. doi:10.1186 / gb-2007-8-8-r171. PMC 2375001. PMID 17708774.
- ^ Chapman, Brad, Instalace Biopython, vyvoláno 11. září 2014
- ^ Chang, Jeff; Chapman, Brad; Friedberg, Iddo; Hamelryck, Thomas; de Hoon, Michiel; Kohout, Peter; Antao, Tiago; Talevich, Eric; Wilczynski, Bartek (29. května 2014), Výukový program a kuchařka pro Biopython, vyvoláno 28. srpna 2014
- ^ Zmasek, Christian M; Zhang, Qing; Ye, Yuzhen; Godzik, Adam (24. října 2007). „Překvapivá složitost sítě předků apoptózy“. Genome Biology. 8 (10): R226. doi:10.1186 / gb-2007-8-10-r226. PMC 2246300. PMID 17958905. Citováno 9. září 2014.
- ^ Talevich, Eric; Invergo, Brandon M; Kohout, Peter JA; Chapman, Brad A (21. srpna 2012). „Bio.Phylo: Jednotná sada nástrojů pro zpracování, analýzu a vizualizaci fylogenetických stromů v Biopythonu“. BMC bioinformatika. 13 (209): 209. doi:10.1186/1471-2105-13-209. PMC 3468381. PMID 22909249. Citováno 9. září 2014.
- ^ "Klebsiella pneumoniae kmen KPS77 plazmid pKPS77, kompletní sekvence". NCBI. Citováno 10. září 2014.
- ^ Pritchard, Leighton; White, Jennifer A; Birch, Paul RJ; Toth, Ian K (březen 2006). „GenomeDiagram: balíček pythonu pro vizualizaci genomických dat ve velkém měřítku“. Bioinformatika. 22 (5): 616–617. doi:10.1093 / bioinformatika / btk021. PMID 16377612.
- ^ Hamelryck, Thomas; Manderick, Bernard (10. května 2003). "Analyzátor souborů PDB a třída struktury implementována v Pythonu". Bioinformatika. 19 (17): 2308–2310. doi:10.1093 / bioinformatika / btg299. PMID 14630660. Citováno 10. září 2014.
- ^ Rousset, François (leden 2008). „GENEPOP'007: kompletní re-implementace softwaru GENEPOP pro Windows a Linux“. Zdroje molekulární ekologie. 8 (1): 103–106. doi:10.1111 / j.1471-8286.2007.01931.x. PMID 21585727. S2CID 25776992.
- ^ Excoffier, Laurent; Foll, Matthieu (1. března 2011). „fastsimcoal: spojitý simulátor genomické diverzity kontinuálního času v libovolně složitých evolučních scénářích“. Bioinformatika. 27 (9): 1332–1334. doi:10.1093 / bioinformatika / btr124. PMID 21398675. Citováno 11. září 2014.