Aktivační energie - Activation energy

v chemie a fyzika, aktivační energie je energie, která musí být poskytnuta sloučeninám, aby došlo k a chemická reakce.[1]Aktivační energie (EA) reakce se měří v joulů na mol (J / mol), kilojoulů na mol (kJ / mol) nebo kilokalorií na mol (kcal / mol).[2] Aktivační energii lze považovat za velikost potenciální bariéra (někdy nazývaná energetická bariéra) oddělující se minima z potenciální energie povrch náležející k počátečnímu a konečnému termodynamický stav. Aby chemická reakce probíhala přiměřenou rychlostí, měla by být teplota systému dostatečně vysoká, aby existoval znatelný počet molekul s translační energií rovnou nebo větší než aktivační energie. Termín Aktivační energie zavedl v roce 1889 švédský vědec Svante Arrhenius.[3]
Jiná použití
I když se méně často používá, aktivační energie platí také pro jaderné reakce[4][5] a různé další fyzikální jevy.[6][7][8][9]
Teplotní závislost a vztah k Arrheniově rovnici
The Arrheniova rovnice poskytuje kvantitativní základ vztahu mezi aktivační energií a rychlostí, jakou reakce probíhá. Z rovnice lze zjistit aktivační energii prostřednictvím vztahu

kde A je preexponenciální faktor pro reakci, R je univerzální plynová konstanta, T je absolutní teplota (obvykle v kelvinů ), a k je koeficient reakční rychlosti. I bez vědomí A, EA lze vyhodnotit z variace koeficientů reakční rychlosti jako funkce teploty (v rámci platnosti Arrheniovy rovnice).
Na pokročilejší úrovni je termín čisté aktivační energie Arrhenius z Arrheniovy rovnice nejlépe považován za experimentálně určený parametr, který udává citlivost reakční rychlosti na teplotu. Proti asociaci této aktivační energie s prahovou bariérou pro elementární reakci existují dvě námitky. Za prvé, často není jasné, zda reakce probíhá v jednom kroku; prahové bariéry, které jsou zprůměrovány ve všech základních krocích, mají malou teoretickou hodnotu. Zadruhé, i když je zkoumaná reakce elementární, spektrum jednotlivých kolizí přispívá k rychlostním konstantám získaným z hromadných („bulb“) experimentů zahrnujících miliardy molekul, s mnoha různými kolizními geometriemi a úhly reaktantů, různými translačními a (případně) vibračními energie - to vše může vést k různým rychlostem mikroskopické reakce.[Citace je zapotřebí ]
Katalyzátory
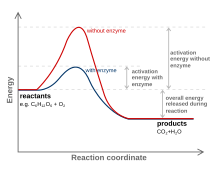


Látka, která mění přechodový stav za účelem snížení aktivační energie, se nazývá a katalyzátor; katalyzátor složený pouze z proteinu a (je-li to relevantní) malomolekulární kofaktory se označuje jako enzym. Katalyzátor zvyšuje rychlost reakce, aniž by byl spotřebován v reakci.[10] Katalyzátor navíc snižuje aktivační energii, ale nemění energie původních reaktantů nebo produktů, a tak nemění rovnováhu.[11] Energie reaktantů a energie produktů spíše zůstávají stejné a jediné aktivační energie je změněn (spuštěn).
Katalyzátor je schopen snížit aktivační energii vytvořením přechodného stavu příznivějším způsobem. Katalyzátory přirozeně vytvářejí „pohodlnější“ přizpůsobení substrátu reakce k přechodu do přechodového stavu. To je možné díky uvolnění energie, ke kterému dochází, když se substrát váže na Aktivní stránky katalyzátoru. Tato energie je známá jako vazebná energie. Po navázání na katalyzátor se substráty účastní mnoha stabilizačních sil, zatímco jsou v aktivním místě (tj. Vodíková vazba, van der Waalsovy síly ). Specifické a příznivé vazby se vyskytují v aktivním místě, dokud se substrát nevytvoří, aby se stal vysokoenergetickým přechodovým stavem. Formování přechodového stavu je s katalyzátorem příznivější, protože příznivé stabilizační interakce uvnitř aktivního místa uvolnění energie. Chemická reakce je schopna snadněji vyrobit molekulu přechodného stavu s vysokou energií, když je stabilizační uložení v aktivním místě katalyzátoru. Vazebná energie reakce je tato energie uvolněná při příznivých interakcích mezi substrátem a katalyzátorem. Uvolněná vazebná energie pomáhá dosáhnout nestabilního přechodového stavu. Reakce jinak bez katalyzátorů potřebují k dosažení přechodového stavu vyšší vstup energie. Nekatalyzované reakce nemají volnou energii dostupnou z interakcí stabilizujících aktivní místo, jako jsou katalytické enzymové reakce.[12]
Vztah s Gibbsovou aktivační energií
V Arrheniova rovnice, termín aktivační energie (EA) se používá k popisu požadované energie dosáhnout přechodový stav a exponenciální vztah k = A exp (-EA/RT) drží. V teorii přechodového stavu je sofistikovanější model vztahu mezi reakčními rychlostmi a přechodovým stavem, povrchně podobný matematický vztah, Eyringova rovnice, se používá k popisu rychlosti reakce: k = (kBT / h) exp (–ΔG‡ / RT). Avšak místo fenomenologického modelování teplotní závislosti reakční rychlosti modeluje Eyringova rovnice jednotlivé elementární kroky reakce. U vícestupňového procesu tedy neexistuje přímý vztah mezi těmito dvěma modely. Přesto jsou funkční formy rovnic Arrhenius a Eyring podobné a pro jednostupňový proces lze mezi parametry Arrhenius a Eyring vyvodit jednoduchou a chemicky významnou shodu.
Místo toho také používat EA, Eyringova rovnice používá koncept Gibbsova energie a symbol ΔG‡ označit Gibbsovu energii aktivace k dosažení přechodový stav. V rovnici kB a h jsou Boltzmannovy a Planckovy konstanty. Ačkoli rovnice vypadají podobně, je důležité si uvědomit, že Gibbsova energie obsahuje entropický termín kromě entalpického. V Arrheniově rovnici je tento entropický člen považován za preexponenciální faktor A. Přesněji, můžeme napsat Gibbsovu volnou energii aktivace ve smyslu entalpie a entropie aktivace: ΔG‡ = ΔH‡ – T ΔS‡. Poté, pro unimolekulární, jednokrokovou reakci, přibližný vztahy EA = ΔH‡ + RT a A = (kBT/h) exp (1 + ΔS‡/R) podržte. Všimněte si však, že ve vlastní Arrheniově teorii A je nezávislá na teplotě, zatímco zde existuje lineární závislost na T. Pro jednostupňový unimolekulární proces, jehož poločas při pokojové teplotě je asi 2 hodiny, ΔG‡ je přibližně 23 kcal / mol. To je také zhruba velikost EA pro reakci, která probíhá několik hodin při teplotě místnosti. Vzhledem k relativně malé velikosti TΔS‡ a RT při běžné teplotě pro většinu reakcí, v nedbalém diskurzu, EA, ΔG‡a ΔH‡ jsou často sjednoceny a všechny se označují jako „aktivační energie“.
Entalpie, entropie a Gibbsova aktivační energie jsou přesněji zapsány jako Δ‡HÓ, Δ‡SÓ a Δ‡GÓ kde o označuje množství vyhodnocené mezi standardní stavy.[13][14] Někteří autoři však o vynechávají, aby zjednodušili notaci.[15][16]
Celková změna volné energie reakce je však nezávislá na aktivační energii. Fyzikální a chemické reakce mohou být buď exergonický nebo endergonický, ale aktivační energie nesouvisí s spontánnost reakce. Celková změna reakční energie se nemění aktivační energií.
Negativní aktivační energie
V některých případech rychlost reakce pokles s rostoucí teplotou. Když sledujete přibližně exponenciální vztah, takže rychlostní konstanta může být stále přizpůsobena Arrheniovi výrazu, vede to k záporné hodnotě EA. Elementární reakce vykazující tyto negativní aktivační energie jsou typicky bezbariérové reakce, při nichž se reakce spoléhá na zachycení molekul v potenciální jamce. Zvýšení teploty vede ke snížené pravděpodobnosti, že se kolidující molekuly zachytí navzájem (přičemž více kolizních kolizí nevede k reakci, protože vyšší hybnost přenáší kolidující částice z potenciální jámy), vyjádřeno jako reakce průřez která klesá s rostoucí teplotou. Taková situace již sama o sobě nevede k přímým interpretacím, jako je výška potenciální bariéry.[17]
Viz také
- Asymptotika aktivační energie
- Chemická kinetika
- Bod požáru
- Střední kinetická teplota
- Kvantové tunelování
- Bezpečnost vodíku
- Výbuch prachu
- Zapalovací svíčka
Reference
- ^ "Aktivační energie". www.chem.fsu.edu. Archivovány od originál dne 2016-12-07. Citováno 2017-01-13.
- ^ Espenson, James (1995). Chemická kinetika a reakční mechanismy. McGraw-Hill. ISBN 0070202605.
- ^ „Activation Energy and the Arrhenius Equation - Introduction Chemistry - 1st Canadian Edition“. opentextbc.ca. Citováno 2018-04-05.
- ^ http://www.physics.ohio-state.edu/~kagan/phy367/Lectures/P367_lec_14.html[úplná citace nutná ]
- ^ „Přednáška XIV“. www.asc.ohio-state.edu. Citováno 2019-03-22.
- ^ Pratt, Thomas H. „Elektrostatické zapalování požárů a výbuchů“ Wiley-AIChE (15. července 1997) Centrum pro bezpečnost chemických procesů[stránka potřebná ]
- ^ Wang, Jenqdaw; Raj, Rishi (1990). „Odhad aktivačních energií pro mezní difúzi z frekvenčně řízeného slinování čistého oxidu hlinitého a oxidu hlinitého dopovaného oxidem zirkoničitým nebo titanickým“. Journal of the American Ceramic Society. 73 (5): 1172. doi:10.1111 / j.1151-2916.1990.tb05175.x.
- ^ Kiraci, A; Yurtseven, H (2012). „Teplotní závislost Ramanovy frekvence, tlumicí konstanty a aktivační energie režimu měkké optiky ve feroelektrickém barnatém titanátu“. Feroelektrika. 432: 14–21. doi:10.1080/00150193.2012.707592. S2CID 121142463.
- ^ Terracciano, Anthony C; De Oliveira, Samuel; Vazquez-Molina, Demetrius; Uribe-Romo, Fernando J; Vasu, Subith S; Orlovskaya, Nina (2017). „Vliv katalyticky aktivního povlaku Ce 0,8 Gd 0,2 O 1,9 na heterogenní spalování methanu v porézní keramice ZrO 2 stabilizované MgO“. Spalování a plamen. 180: 32–39. doi:10.1016 / j.combustflame.2017.02.019.
- ^ „General Chemistry Online: FAQ: Chemické změny: Jaké jsou příklady reakcí zahrnujících katalyzátory?“. antoine.frostburg.edu. Citováno 2017-01-13.
- ^ Bui, Matthew. „Arrhenův zákon: aktivační energie“. Chemistry LibreTexts. UC Davis. Citováno 17. února 2017.
- ^ Berg, Jeremy (2019). Biochemistry - Deváté vydání. New York, NY: WH Freeman and Company. 240–244. ISBN 978-1-319-11467-1.
- ^ „Entalpie aktivace“. Zlatá kniha IUPAC (2. vydání, online verze). IUPAC (Mezinárodní unie pro čistou a aplikovanou chemii). 2019. Citováno 10. května 2020.
- ^ Steinfeld, Jeffrey I .; Francisco, Joseph S .; Hase, William L. (1999). Chemická kinetika a dynamika (2. vyd.). Prentice Hall. p. 301. ISBN 0-13-737123-3.
- ^ Atkins, Peter; de Paula, Julio (2006). Atkinsova fyzikální chemie (8. vydání). WH Freeman. p.883. ISBN 0-7167-8759-8.
... ale vynecháme standardní státní znak, abychom zabránili přetížení notace.
- ^ Laidler, Keith J .; Meiser, John H. (1982). Fyzikální chemie. Benjamin / Cummings. p. 381. ISBN 0-8053-5682-7.
- ^ Mozurkewich, Michael; Benson, Sidney (1984). „Negativní aktivační energie a zakřivené Arrheniovy grafy. 1. Teorie reakcí nad potenciálními jamkami“. J. Phys. Chem. 88 (25): 6429–6435. doi:10.1021 / j150669a073.