Virtuální karyotyp - Virtual karyotype
Virtuální karyotyp je digitální informace odrážející a karyotyp, který je výsledkem analýzy krátkých sekvencí DNA ze specifických lokusů po celém genomu, které jsou izolovány a vyjmenovány.[1] Detekuje genomické kopírovat varianty počtu při vyšším rozlišení na úrovni než u konvenčních karyotypů nebo na základě chromozomů komparativní genomová hybridizace (CGH).[2] Hlavní metody používané při vytváření virtuálních karyotypů jsou genomová hybridizace srovnávající pole a Pole SNP.
Pozadí
A karyotyp (Obr. 1) je charakteristika chromozóm doplněk a eukaryot druh.[3][4] Karyotyp je obvykle prezentován jako obraz chromozomů z jedné buňky uspořádané od největší (chromozom 1) po nejmenší (chromozom 22), přičemž poslední jsou zobrazeny pohlavní chromozomy (X a Y). Historicky byly karyotypy získány barvením buněk poté, co byly chemicky zastaveny během dělení buněk. Karyotypy se používají již několik desetiletí k identifikaci chromozomálních abnormalit v zárodečných i rakovinových buňkách. Konvenční karyotypy mohou vyhodnotit změny ve struktuře a počtu chromozomů v celém genomu, ale rozlišení je relativně hrubé, s detekčním limitem 5-10 Mb.

Metoda
V poslední době platformy pro generování karyotypů s vysokým rozlišením in silico z narušené DNA se objevily, jako např pole komparativní genomová hybridizace (arrayCGH) a Pole SNP. Koncepčně jsou pole složena ze stovek až milionů sond, které se doplňují k oblasti zájmu v genomu. Narušená DNA z testovaného vzorku je fragmentována, označena a hybridizována s maticí. Intenzity hybridizačního signálu pro každou sondu používá specializovaný software ke generování log2ratio testu / normálu pro každou sondu na poli. Znát adresu každé sondy na poli a adresu každé sondy v genomu, software seřadí sondy v chromozomálním pořadí a rekonstruuje genom in silico (Obr. 2 a 3).
Virtuální karyotypy mají dramaticky vyšší rozlišení než konvenční cytogenetika. Skutečné rozlišení bude záviset na hustotě sond na poli. V současné době Affymetrix SNP6.0 je komerčně dostupné pole s nejvyšší hustotou pro aplikace virtuálních karyotypů. Obsahuje 1,8 milionu polymorfních a nepolymorfních markerů pro praktické rozlišení 10–20 kB - přibližně o velikosti genu. To je přibližně 1000krát větší rozlišení než karyotypy získané z konvenční cytogenetiky.
Virtuální karyotypy lze provádět na zárodečných vzorcích pro ústavní poruchy,[5][6] a klinické testování je k dispozici v desítkách laboratoří certifikovaných podle CLIA (genetests.org ). Virtuální karyotypizaci lze provést také na čerstvých nebo ve formalínu fixovaných parafínových nádorech.[7][8][9] Mezi laboratoře s certifikátem CLIA, které nabízejí testování na nádory, patří Creighton Medical Laboratories (čerstvé a parafínové vzorky nádoru) a Molekulární diagnostika CombiMatrix (čerstvé vzorky nádoru).


Různé platformy pro virtuální karyotyping
Karyotypizaci založenou na poli lze provést pomocí několika různých platforem, laboratorně vyvinutých i komerčních. Samotná pole mohou být celá v genomu (sondy rozmístěné po celém genomu) nebo cílená (sondy pro genomické oblasti, o nichž je známo, že se účastní konkrétního onemocnění), nebo kombinace obou. Pole používaná pro karyotypování mohou dále používat nepolymorfní sondy, polymorfní sondy (tj. Obsahující SNP) nebo kombinaci obou. Nepolymorfní sondy mohou poskytovat pouze informace o počtu kopií, zatímco pole SNP mohou poskytovat počet kopií i stav ztráty heterozygotnosti (LOH) v jednom testu. Typy sond používané pro nepolymorfní pole zahrnují cDNA, BAC klony (např. BlueGnome ) a oligonukleotidy (např. Agilent, Santa Clara, CA, USA nebo Nimblegen, Madison, WI, USA). Komerčně dostupné oligonukleotidové pole SNP mohou být v pevné fázi (Affymetrix, Santa Clara, CA, USA) nebo na bázi korálků (Illumina, SanDiego, CA, USA). Navzdory rozmanitosti platforem nakonec všichni používají genomovou DNA z narušených buněk k vytvoření karyotypu s vysokým rozlišením in silico. Konečný produkt ještě nemá konzistentní název a byl nazýván virtuální karyotyping,[8][10] digitální karyotypizace,[11] molekulární alelokaryotypizace,[12] a molekulární karyotypizace.[13] Mezi další výrazy používané k popisu polí používaných pro karyotypizaci patří SOMA (SNP oligonukleotidové mikropole)[14] a CMA (chromosomová microarray).[15][16] Někteří považují všechny platformy za určitý typ pole komparativní genomová hybridizace (arrayCGH), zatímco jiní si tento termín vyhrazují pro dvoubarevné metody, a ještě další oddělují pole SNP, protože generují více a odlišných informací než dvoubarevné metody arrayCGH.
Aplikace
Zjištění změn počtu kopií
Změny počtu kopií lze vidět ve vzorcích zárodečné linie i nádoru. Změny počtu kopií lze zjistit pomocí polí s nepolymorfními sondami, jako je například arrayCGH, a pomocí polí založených na SNP. Lidské bytosti jsou diploidní, takže u nepohlavních chromozomů je normální počet kopií vždy dva.
- Odstranění: A vymazání je ztráta genetického materiálu. Delece může být heterozygotní (počet kopií 1) nebo homozygotní (počet kopií 0, nullizomie). Mikrodeleční syndromy jsou příklady konstitučních poruch způsobených malými delecemi v zárodečné DNA. Delece v nádorových buňkách mohou představovat inaktivaci tumor-supresorového genu a mohou mít diagnostické, prognostické nebo terapeutické důsledky.
- Zisky: Zisk počtu kopií představuje zisk genetického materiálu. Pokud je zisk pouze jednou další kopií segmentu DNA, lze jej nazvat a zdvojení (Obr. 4). Pokud existuje ještě jedna kopie celého chromozomu, lze jej nazvat a trizomie. Zisk počtu kopií ve vzorcích zárodečné linie může být spojen s onemocněním nebo může být neškodný varianta počtu kopií. Jsou-li pozorovány v nádorových buňkách, mohou mít diagnostické, prognostické nebo terapeutické důsledky.
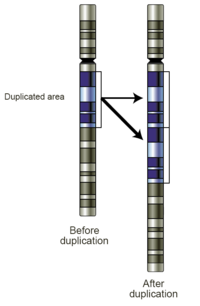
- Zesílení: Technicky vzato zesílení je typ nárůstu počtu kopií, ve kterém je počet kopií> 10. V kontextu biologie rakoviny jsou často pozorovány amplifikace onkogeny. To by mohlo naznačovat horší prognózu, pomoci kategorizovat nádor nebo indikovat vhodnost léku. Příkladem způsobilosti k léčbě je amplifikace Her2Neu a Herceptin a je poskytnut obraz amplifikace Her2Neu detekovaný virtuálním karyotypováním pole SNP (obr. 5).
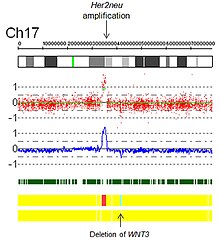 Obr. 5. Zesílení Her2 pomocí virtuálního karyotypu pole SNP.
Obr. 5. Zesílení Her2 pomocí virtuálního karyotypu pole SNP.

Ztráta heterozygotnosti (LOH), autozygotních segmentů a uniparentální disomie
Autozygotní segmenty a uniparental disomy (UPD) jsou diploidní / „kopii neutrální“ genetické nálezy, a proto jsou detekovatelné pouze poli založenými na SNP. Zobrazí se oba autozygotní segmenty a UPD ztráta heterozygotnosti (LOH) s kopií číslo dvě pomocí karyotypizace pole SNP. Termín Runs of Homozgygosity (ROH), je obecný termín, který lze použít buď pro autozygotní segmenty, nebo pro UPD.
- Autozygotní segment: Autozygotní segment je bi-rodičovský a je vidět pouze v zárodečné linii. Jsou to prodloužené série homozygotních markerů v genomu a vyskytují se, když jsou identické haplotyp blok je zděděn od obou rodičů. Také se jim říká „identické sestupem "(IBD) segmenty a lze je použít pro mapování homozygotnosti.[17][18]
- Uniparental Disomy: UPD nastává, když jsou obě kopie genu nebo genomové oblasti zděděny od stejného rodiče. To je uniparental, na rozdíl od autozygotních segmentů, které jsou bi-parentální. Pokud jsou přítomny v zárodečné linii, mohou být neškodné nebo spojené s onemocněním, jako je například Prader-Willi nebo Angelmanovy syndromy. Na rozdíl od autozygotnosti se UPD může vyvinout v nádorových buňkách, což se v literatuře označuje jako získaný UPD nebo kopírování neutrálního LOH (obr. 6). Získaná UPD je zcela běžná u hematologických i solidních nádorů a uvádí se, že tvoří 20 až 80% LOH pozorovaných u lidských nádorů.[19][20][21][22] Získaný UPD může sloužit jako druhý zásah v Knudson Two Hit Hypotéza tumorigeneze, a tak může být biologickým ekvivalentem delece.[23] Protože tento typ léze nelze detekovat pomocí arrayCGH, FISH nebo konvenční cytogenetiky, jsou pro virtuální karyotypizaci nádorů preferována pole založená na SNP.
 Obr. 6. Zkopírujte neutrální LOH / uniparentalní disomii
Obr. 6. Zkopírujte neutrální LOH / uniparentalní disomii


Obrázek 7 je virtuální karyotyp pole SNP z kolorektálního karcinomu, který demonstruje delece, zisky, amplifikace a získaný UPD (copy neutral LOH).
Příklady klinických aplikací rakoviny
Virtuální karyotyp lze generovat téměř z jakéhokoli nádoru, ale klinický význam identifikovaných genomových aberací je pro každý typ nádoru odlišný. Klinická užitečnost se liší a vhodnost nejlépe určí onkolog nebo patolog po konzultaci s ředitelem laboratoře provádějící virtuální karyotyp. Níže jsou uvedeny příklady typů rakoviny, u kterých jsou dobře známy klinické důsledky specifických genomových aberací. Tento seznam je reprezentativní, není vyčerpávající. Webové stránky Cytogenetics Laboratory ve Wisconsin State Laboratory of Hygiene obsahují další příklady klinicky relevantních genetických změn, které lze snadno zjistit pomocí virtuálního karyotypizace.[1]
Neuroblastom
Na základě série 493 neuroblastom vzorků, bylo hlášeno, že celkový genomický vzorec, jak je testován karyotypováním založeným na poli, je prediktorem výsledku v neuroblastomu:[24]
- Nádory vykazující výhradně změny počtu kopií celého chromozomu byly spojeny s vynikajícím přežitím.
- Nádory s jakýmkoli druhem změn počtu kopií segmentových chromozomů byly spojeny s vysokým rizikem relapsu.
- V nádorech vykazujících segmentové změny byly dalšími nezávislými prediktory snížení celkového přežití amplifikace MYCN, delece 1p a 11q a 1q zisk.
Dřívější publikace kategorizovaly neuroblastomy do tří hlavních podtypů na základě cytogenetických profilů:[25]
- Podtyp 1: příznivý neuroblastom s téměř triploidií a převahou numerických zisků a ztrát, většinou představujících nemetastatické stupně NB 1, 2 a 4S.
- Podtypy 2A a 2B: nalezené v nepříznivém rozšířeném neuroblastomu, stupně 3 a 4, se ztrátou 11q a ziskem 17q bez amplifikace MYCN (podtyp 2A) nebo s amplifikací MYCN často společně s delecemi 1p a ziskem 17q (podtyp 2B).
Wilmsův nádor
Ztráta heterozygosity specifické pro nádor (LOH) pro chromozomy 1p a 16q identifikuje podmnožinu Wilmsův nádor pacienti, u kterých je významně zvýšené riziko relapsu a úmrtí. LOH pro tyto chromozomální oblasti lze nyní použít jako nezávislý prognostický faktor společně se stádiem onemocnění k zaměření intenzity léčby na riziko selhání léčby.[26][27]
Karcinom ledvin
Renální epiteliální neoplazmy mají charakteristické cytogenetické aberace, které mohou pomoci při klasifikaci.[28] Viz také Atlas genetiky a cytogenetiky v onkologii a hematologii.
- Jasný buněčný karcinom: ztráta 3p
- Papilární karcinom: trizomie 7 a 17
- Chromofobní karcinom: hypodiploid se ztrátou chromozomů 1, 2, 6, 10, 13, 17, 21
Karyotypizaci založenou na poli lze použít k identifikaci charakteristických chromozomálních aberací u nádorů ledvin s náročnou morfologií.[8][10] Karyotypizace založená na polích funguje dobře u nádorů zalitých v parafinu[29] a je vhodný pro rutinní klinické použití.
Kromě toho nedávná literatura naznačuje, že určité chromozomální aberace jsou spojeny s výsledkem u konkrétních podtypů renálních epitelových nádorů.[30]
Čistý buněčný renální karcinom: del 9p a del 14q jsou špatné prognostické ukazatele.[31][32]
Papilární karcinom ledvinových buněk: duplikace 1q znamená fatální progresi.[33]
Chronická lymfocytární leukémie
Karyotypizace založená na poli je nákladově efektivní alternativou k FISH pro detekci chromozomálních abnormalit v chronická lymfocytární leukémie (CLL). Několik klinických studií validace prokázalo> 95% shodu se standardním panelem CLL FISH.[12][34][35][36][37] Mnoho studií využívajících karyotypizaci založenou na poli navíc identifikovalo „atypické delece“ vynechané standardními sondami FISH a získalo uniparentální disomii v klíčových lokusech pro prognostické riziko u CLL.[38][39]
V buňkách CLL jsou rozpoznány čtyři hlavní genetické aberace, které mají zásadní vliv na chování nemoci.[40]
- Obzvláště škodlivé jsou delece části krátkého ramene chromozomu 17 (del 17p), které cílí na p53. Pacienti s touto abnormalitou mají významně krátký interval před tím, než vyžadují terapii a kratší přežití. Tato abnormalita se vyskytuje u 5–10% pacientů s CLL.
- Delece dlouhého ramene na chromozomu 11 (del 11q) jsou také nepříznivé, i když ne v míře pozorované u del 17p. Abnormalita se zaměřuje na gen ATM a vyskytuje se zřídka v CLL (5–10%).
- Trizomie 12, další chromozom 12, je relativně častým nálezem vyskytujícím se u 20–25% pacientů a poskytuje střední prognózu.
- Delece 13q14 (del 13q14) je nejčastější abnormalitou u CLL u zhruba 50% pacientů s buňkami obsahujícími tento defekt. Když je del 13q14 viděn izolovaně, mají pacienti nejlepší prognózu a většina z nich bude žít mnoho let, dokonce i desetiletí, bez nutnosti terapie.
Mnohočetný myelom
Avet-Loiseau a kol. v časopise Journal of Clinical Oncology použil karyotypizaci pole SNP 192 mnohočetný myelom (MM) vzorky k identifikaci genetických lézí spojených s prognózou, které byly poté validovány v samostatné kohortě (n = 273).[41] U MM nedostatek proliferačního klonu činí konvenční cytogenetiku informativní pouze v ~ 30% případů. Panely FISH jsou užitečné u MM, ale standardní panely by nezjistily několik klíčových genetických abnormalit hlášených v této studii.
- Virtuální karyotypizace identifikovala chromozomální abnormality u 98% případů MM
- del (12p13.31) je nezávislý nepříznivý marker
- amp (5q31.1) je příznivý marker
- Prognostický dopad amp (5q31.1) převyšuje účinek hyperdiploidie a také identifikuje pacienty, kteří velmi těží z léčby vysokými dávkami.
Karyotypizace založená na poli nedokáže detekovat vyvážené translokace, jako je t (4; 14) pozorované u ~ 15% MM. FISH pro tuto translokaci by se proto měl provádět také při použití polí SNP k detekci změn počtu kopií genomu s prognostickým významem u MM.
Medulloblastom
Pole založené na karyotypizaci 260 meduloblastomy Pfister S, et al. vedly k následujícím klinickým podskupinám založeným na cytogenetických profilech:[42]
- Špatná prognóza: zisk 6q nebo zesílení MYC nebo MYCN
- Střední: zisk 17q nebo i (17q) bez zisku 6q nebo zesílení MYC nebo MYCN
- Vynikající prognóza: 6q a 17q vyvážené nebo 6q delece
Oligodendroglioma
Společná delece 1p / 19q je považována za „genetický podpis“ oligodendroglioma. Alelické ztráty na 1p a 19q, ať už samostatně nebo v kombinaci, jsou častější u klasických oligodendrogliomů než u astrocytomů nebo oligoastrocytomů.[43] V jedné studii vykázaly klasické oligodendrogliomy ztrátu 1p u 35 ze 42 (83%) případů, 19q ztrátu u 28 z 39 (72%) a tyto byly kombinovány ve 27 z 39 (69%) případů; nebyl zjištěn žádný významný rozdíl ve ztrátě stavu heterozygotnosti 1p / 19q mezi nízko kvalitními a anaplastickými oligodendrogliomy.[43] Ko-delece 1p / 19q byla korelována jak s chemosenzitivitou, tak se zlepšenou prognózou u oligodendrogliomů.[44][45] Většina větších středisek pro léčbu rakoviny běžně kontroluje odstranění 1p / 19q jako součást patologie zpráva pro oligodendrogliomy. Stav lokusů 1p / 19q lze zjistit pomocí FISH nebo virtuálního karyotypování. Výhodou virtuálního karyotypizace je hodnocení celého genomu v jednom testu, jakož i lokusů 1p / 19q. To umožňuje posouzení dalších klíčových lokusů v gliových nádorech, jako je stav počtu kopií EGFR a TP53.
Zatímco prognostická relevance delecí 1p a 19q je dobře zavedena pro anaplastické oligodendrogliomy a smíšené oligoastrocytomy, prognostická relevance delecí pro gliomy nízkého stupně je kontroverznější. Pokud jde o gliomy nízkého stupně, nedávná studie také naznačuje, že ko-delece 1p / 19q může být spojena s translokací (1; 19) (q10; p10), která je, stejně jako kombinovaná delece 1p / 19q, spojena s vynikající celkové přežití a přežití bez progrese u pacientů s nízkým stupněm gliomu.[46] Oligodendrogliomy vykazují jen zřídka mutace v genu p53, což je na rozdíl od jiných gliomů.[47] Receptor epidermálního růstového faktoru amplifikace a celá kodifikace 1p / 19q se vzájemně vylučují a predikují zcela odlišné výsledky, přičemž amplifikace EGFR předpovídá špatnou prognózu.[48]
Glioblastom
Yin a kol.[49] studoval 55 glioblastom a 6 GBM buněčné linie využívající karyotypizaci pole SNP. Získaná UPD byla identifikována na 17p v 13/61 případech. Významně zkrácená doba přežití byla nalezena u pacientů s delecí 13q14 (RB) nebo delecí / získanou UPD 17p13.1 (p53). Dohromady tyto výsledky naznačují, že tato technika je rychlá, robustní a levná metoda pro profilování abnormalit v GBM v celém genomu. Vzhledem k tomu, že karyotypizaci pole SNP lze provádět na nádorech zalitých v parafinu, je to atraktivní volba, když nádorové buňky nerostou v kultuře pro metafázovou cytogenetiku nebo když touha po karyotypizaci nastane po fixaci vzorku.
Důležitost detekce získaných UPD (copy neutral LOH) v glioblastomu:
- Z pacientů s abnormalitou 17p bylo ~ 50% delecí a ~ 50% bylo aUPD
- Jak 17p del, tak 17p UPD byly spojeny s horším výsledkem.
- 9/13 mělo homozygotní mutace TP53 pod 17p UPD.
Navíc v případech s nejistým stupněm podle morfologie může genomické profilování pomoci při diagnostice.
- Souběžný zisk 7 a ztráta 10 je pro GBM v podstatě patognomický[50]
- Amplifikace EGFR, ztráta PTEN (na 10q) a ztráta p16 (na 9p) se vyskytují téměř výlučně v glioblastomu a mohou poskytnout prostředky k rozlišení anaplastického astrocytomu od glioblastomu.[51]
Akutní lymfoblastická leukémie
Cytogenetika, studium charakteristických velkých změn v chromozomy z rakovinné buňky, byl stále více uznáván jako důležitý prediktor výsledku v akutní lymfoblastická leukémie (VŠECHNO).[52]
Pozn .: Vyvážené translokace nelze detekovat pomocí karyotypizace založeného na poli (viz Omezení níže).
Některé cytogenetické podtypy mají horší prognózu než jiné. Tyto zahrnují:
- Translokace mezi chromozomy 9 a 22, známé jako Philadelphia chromozom, se vyskytuje asi u 20% dospělých a 5% u pediatrických případů ALL.
- K translokaci mezi chromozomy 4 a 11 dochází přibližně ve 4% případů a je nejčastější u kojenců mladších 12 měsíců.
- Ne všechny translokace chromozomů mají horší prognózu. Některé translokace jsou relativně příznivé. Například hyperdiploidie (> 50 chromozomů) je dobrým prognostickým faktorem.
- Posouzení změn počtu kopií v celém genomu lze provést konvenční cytogenetikou nebo virtuální karyotypizací. Virtuální karyotypizace pole SNP dokáže detekovat změny počtu kopií a stav LOH, zatímco arrayCGH dokáže detekovat pouze změny počtu kopií. Zkopírujte neutrální LOH (získaná uniparentální disomie) byla hlášena u klíčových lokusů u ALL, jako je gen CDKN2A v 9p, které mají prognostický význam.[53][54][55] Virtuální karyotypizace pole SNP může snadno detekovat kopii neutrálního LOH. Pole CGH, FISH a konvenční cytogenetika nedokážou detekovat kopii neutrální LOH.
| Cytogenetická změna | Kategorie rizika |
|---|---|
| Philadelphia chromozom | Špatná prognóza |
| t (4; 11) (q21; q23) | Špatná prognóza |
| t (8; 14) (q24,1; q32) | Špatná prognóza |
| Komplex karyotyp (více než čtyři abnormality) | Špatná prognóza |
| Nízký hypodiploidie nebo blízko triploidie | Špatná prognóza |
| Vysoký hyperdiploidie | Dobrá prognóza |
| del (9p) | Dobrá prognóza |
Korelace prognózy s cytogenetickým nálezem kostní dřeně u akutní lymfoblastické leukémie
| Prognóza | Cytogenetické nálezy |
|---|---|
| Příznivý | Hyperdiploidie> 50; t (12; 21) |
| středně pokročilí | Hyperdioloidie 47-50; Normální (diploidie); del (6q); Přeskupení 8q24 |
| Nepříznivý | Hypodiploidie - téměř haploidie; Téměř tetraploidie; del (17p); t (9; 22); t (11q23) |
Za neklasifikovanou ALL se považuje střední prognóza.[56]
Myelodysplastický syndrom
Myelodysplastický syndrom (MDS) má pozoruhodnou klinickou, morfologickou a genetickou heterogenitu. Cytogenetika hraje rozhodující roli v mezinárodním prognostickém skórovacím systému (IPSS) Světové zdravotnické organizace pro MDS.[57][58]
- Dobrá prognóza: normální karyotyp, izolovaný del (5q), izolovaný del (20q), -Y
- Špatná prognóza: komplexní abnormality (tj.> = 3 abnormality), −7 nebo del (7q)
- Intermediate Prognosis: all other abnormalities, including trisomy 8 and del (11q)
Při srovnání metafázové cytogenetiky, panelu FISH a karyotypizace pole SNP pro MDS bylo zjištěno, že každá technika poskytuje podobný diagnostický výtěžek. Žádná jednotlivá metoda nezjistila všechny vady a míry detekce se zlepšily o ~ 5%, když byly použity všechny tři metody.[59]
Získaná UPD, která není detekovatelná FISH ani cytogenetikou, byla hlášena na několika klíčových lokusech v MDS pomocí karyotypizace pole SNP, včetně delece 7 / 7q.[60][61]
Myeloproliferativní neoplazmy / myeloproliferativní poruchy
Philadelphia chromozom - negativní myeloproliferativní neoplazmy (MPN) včetně polycythemia vera, esenciální trombocytémie a primární myelofibrózy vykazují inherentní tendenci k transformaci na leukémii (MPN-blastová fáze), která je doprovázena získáváním dalších genomových lézí. Ve studii 159 případů[62] Analýza SNP-pole dokázala zachytit prakticky všechny cytogenetické abnormality a odhalit další léze s potenciálně důležitými klinickými důsledky.
- Počet genomových změn byl v blastové fázi více než 2 až 3krát vyšší než v chronické fázi onemocnění.
- Delece 17p (TP53) byla významně spojena s předchozí expozicí hydroxymočovině a také s komplexním karyotypem ve vzorcích s MPN-blastickou krizí. Delece i 17p kopie neutrálního LOH byly spojeny s komplexním karyotypem, špatným prognostickým markerem u myeloidních malignit. Kopie neutrální LOH (získaná UPD) je snadno detekovatelná karyotypem pole SNP, ale ne cytogenetikou, FISH nebo CGH pole.
- Pacienti s blastovou fází se ztrátou chromozomálního materiálu na 7q vykazovali špatné přežití. Je známo, že ztráta 7q je prediktivní pro rychlou progresi a špatnou odpověď v terapii AML. Pacienti s MPN-blastovou fází s cytogeneticky nedetekovatelnou 7q kopií neutrálního LOH měli srovnatelnou míru přežití s pacienty se 7 / 7q v jejich leukemických buňkách.
- 9p kopie neutrální-LOH s homozygotní mutací JAK2 byla také spojena s horším výsledkem MPN-blastické krize ve srovnání s pacienty s heterozygotní JAK2V617F nebo divokým typem JAK2. Na rozdíl od LOH na 17p byl prognostický dopad 9pCNN-LOH nezávislý na zavedených rizikových faktorech, jako je 7 / 7q, 5q nebo komplexní karyotyp.
Kolorektální karcinom
Identifikace biomarkerů v EU kolorektální karcinom je zvláště důležitá pro pacienty s onemocněním stádia II, kde se u méně než 20% vyskytuje recidiva nádoru. 18q LOH je zavedený biomarker spojený s vysokým rizikem recidivy nádoru ve stadiu II rakoviny tlustého střeva.[63] Obrázek 7 ukazuje karyotyp pole SNP kolorektálního karcinomu (pohled na celý genom).
Kolorektální rakoviny jsou klasifikovány do specifických fenotypů nádoru na základě molekulárních profilů[63] které lze integrovat s výsledky dalších pomocných testů, jako je testování nestability mikrosatelitů, IHC a stav mutace KRAS:
- Chromozomální nestabilita (CIN), která má alelickou nerovnováhu na řadě chromozomálních lokusů, včetně 5q, 8p, 17p a 18q (obr. 7).
- Mikrosatelitní nestabilita (MSI), které mají tendenci mít diploidní karyotypy.
Maligní rhabdoidní nádory
Maligní rhabdoidní nádory jsou vzácné, vysoce agresivní novotvary vyskytující se nejčastěji u kojenců a malých dětí. Vzhledem k jejich heterogenním histologickým vlastnostem může být diagnostika často obtížná a může dojít k nesprávné klasifikaci. V těchto nádorech funguje gen INI1 (SMARCB1) na chromozomu 22q jako klasický tumor supresorový gen. K deaktivaci INI1 může dojít delecí, mutací nebo získanou UPD.[64]
V nedávné studii[64] Karyotypizace pole SNP identifikovala delece nebo LOH 22q u 49/51 rhabdoidních nádorů. Z nich bylo 14 kopií neutrálních LOH (nebo získaných UPD), které jsou detekovatelné pomocí karyotypizace pole SNP, ale nikoli pomocí FISH, cytogenetiky nebo arrayCGH. MLPA detekovala jedinou exonovou homozygotní deleci v jednom vzorku, který byl pod rozlišením pole SNP.
Karyotypizaci pole SNP lze použít k rozlišení například meduloblastomu s izochromozomem 17q od primárního rhabdoidního tumoru se ztrátou 22q11.2. Pokud je to indikováno, může být použita molekulární analýza INI1 pomocí MLPA a přímé sekvenování. Jakmile jsou nalezeny změny spojené s nádorem, lze provést analýzu zárodečné DNA od pacienta a rodičů, aby se vyloučila dědičná nebo de novo zárodečná mutace nebo delece INI1, aby bylo možné provést odpovídající hodnocení rizika recidivy.[64]
Uvealní melanom
Nejdůležitější genetická alterace spojená se špatnou prognózou v roce 2006 uveální melanom je ztráta celé kopie Chromozom 3 (Monosomie 3), který silně koreluje s metastatickým rozšířením.[65] Zisky na chromozomech 6 a 8 se často používají k upřesnění prediktivní hodnoty obrazovky Monosomy 3, přičemž zisk 6p naznačuje lepší prognózu a zisk 8q naznačuje horší prognózu nepořádek 3 nádory.[66] Ve vzácných případech mohou nádory monosomie 3 duplikovat zbývající kopii chromozomu a vrátit se do disomického stavu označovaného jako isodisomie.[67] Isodisomie 3 je prognosticky ekvivalentní monosomii 3 a obě lze detekovat pomocí testů na chromozom 3 ztráta heterozygotnosti.[68]
Omezení
Na rozdíl od karyotypů získaných z konvenční cytogenetiky jsou virtuální karyotypy rekonstruovány počítačovými programy pomocí signálů získaných z narušen DNA. Počítačový program v podstatě opraví translokace, když seřadí signály v chromozomálním pořadí. Virtuální karyotypy proto nemohou detekovat vyvážené translokace a inverze. Mohou také detekovat genetické aberace pouze v oblastech genomu, které jsou reprezentovány sondami v poli. Virtuální karyotypy navíc generují a relativní počet kopií normalizovaný proti diploidnímu genomu, takže tetraploidní genomy budou kondenzovány do diploidního prostoru, pokud nebude provedena renormalizace. Renormalizace vyžaduje test na pomocných buňkách, jako je FISH, pokud jeden používá arrayCGH. U karyotypů získaných z polí založených na SNP lze tetraploidii často odvodit z udržování heterozygotnosti v oblasti zjevné ztráty počtu kopií.[22] Virtuální karyotypy nemusí detekovat mozaiku nízké úrovně nebo malé subklony, protože přítomnost normálních buněk ve vzorku tlumí signál z abnormálního klonu. Přesný bod selhání, pokud jde o minimální procento neoplastických buněk, bude záviset na konkrétní použité platformě a algoritmech. Mnoho softwarových programů pro analýzu počtu kopií používaných ke generování karyotypů založených na matici bude váhat s méně než 25–30% nádorových / abnormálních buněk ve vzorku. V onkologických aplikacích však toto omezení lze minimalizovat strategiemi obohacení nádoru a softwarem optimalizovaným pro použití s onkologickými vzorky. Algoritmy analýzy se rychle vyvíjejí a některé jsou dokonce navrženy tak, aby prospívaly „normální kontaminaci klonem“,[69] Předpokládá se tedy, že toto omezení se bude i nadále rozplývat.
Viz také
- ROZLUŠTIT, Databáze chromozomální nerovnováhy a fenotypu u lidí využívajících prostředky Ensembl
Reference
- ^ Digitální karyotypizace - Wang a kol., 10.1073 / pnas.202610899 - Sborník Národní akademie věd
- ^ Shinawi M, Cheung SW (2008). "Pole CGH a jeho klinické aplikace". Drug Discov dnes. 13 (17–18): 760–70. doi:10.1016 / j.drudis.2008.06.007. PMID 18617013.
- ^ Bílý M.J.D. 1973. Chromozomy. 6. vydání, Chapman & Hall, Londýn. p28
- ^ Stebbins GL 1950. Variace a evoluce v rostlinách. Kapitola XII: Karyotyp. Columbia University Press N.Y.
- ^ Shaffer LG, Bejjani B (2006). "Lékařské aplikace pole CGH a transformace klinické cytogenetiky". Cytogenet. Genome Res. 115 (3–4): 303–9. doi:10.1159/000095928. PMID 17124414.
- ^ Edelmann L, Hirschhorn K (leden 2009). "Klinická užitečnost pole CGH pro detekci chromozomální nerovnováhy spojené s mentální retardací a mnohočetnými vrozenými anomáliemi". Annals of the New York Academy of Sciences. 1151 (1): 157–66. doi:10.1111 / j.1749-6632.2008.03610.x. PMID 19154522.
- ^ Dutt A, Beroukhim R (leden 2007). "Jednorázová nukleotidová polymorfní analýza pole rakoviny". Aktuální názor na onkologii. 19 (1): 43–9. doi:10.1097 / CCO.0b013e328011a8c1. PMID 17133111.
- ^ A b C Hagenkord JM; Parwani AV; Lyons-Weiler MA; Alvarez K; Amato R; Gatalica Z; Gonzalez-Berjon JM; Peterson L; Dhir R; Monzon FA (listopad 2008). „Virtuální karyotypizace pomocí SNP mikročipů snižuje nejistotu v diagnostice renálních epiteliálních nádorů“. Diagn Pathol. 3 (1): 44. doi:10.1186/1746-1596-3-44. PMC 2588560. PMID 18990225.
- ^ Beaudet AL, Belmont J (2008). „Diagnostika DNA založená na poli: nechte revoluci začít“. Annu Rev Med. 59 (1): 113–29. doi:10.1146 / annurev.med.59.012907.101800. PMID 17961075.
- ^ A b Monzon FA; Hagenkord JM; Lyons-Weiler MA; Balani JP; Parwani AV; Sciulli CM; Li J; Chandran UR; Bastacky SI; Dhir R (květen 2008). „Pole celého genomu SNP jako potenciální diagnostický nástroj pro detekci charakteristických chromozomálních aberací v renálních epiteliálních nádorech“. Mod Pathol. 21 (5): 599–608. doi:10.1038 / modpathol.2008.20. PMID 18246049.
- ^ Leary RJ; Lin JC; Cummins J; Boca S; Dřevo LD; Parsons DW; Jones S; Sjöblom T; Park BH; Parsons R; Willis J; Dawson D; Willson JK; Nikolskaya T; Nikolsky Y; Kopelovich L; Papadopoulos N; Pennacchio LA; Wang TL; Markowitz SD; Parmigiani G; Kinzler KW; Vogelstein B; Velculescu VE (2008). „Integrovaná analýza homozygotních delecí, fokálních amplifikací a alterací sekvencí u karcinomů prsu a kolorektálního karcinomu“. Proc Natl Acad Sci U S A. 105 (42): 16224–9. doi:10.1073 / pnas.0808041105. PMC 2571022. PMID 18852474.
- ^ A b Lehmann S; Ogawa S; Raynaud SD; Sanada M; Nannya Y; Ticchioni M; Bastard C; Kawamata N; Koeffler HP (březen 2008). "Molekulární allelokaryotypizace časného stadia, neléčené chronické lymfocytární leukémie". Rakovina. 112 (6): 1296–305. doi:10.1002 / cncr.23270. PMID 18246537.
- ^ Vermeesch JR; Fiegler H; de Leeuw N; Szuhai K; Schoumans J; Ciccone R; Speleman F; Rauch A; Clayton-Smith J; Van Ravenswaaij C; Sanlaville D; Patsalis PC; Firth H; Devriendt K; Zuffardi O (listopad 2007). „Pokyny pro molekulární karyotypizaci v konstituční genetické diagnostice“. Eur J Hum Genet. 15 (11): 1105–14. doi:10.1038 / sj.ejhg.5201896. PMID 17637806.
- ^ Kulharya AS, Flannery DB, Norris K, Lovell C, Levy B, Velagaleti G (září 2008). "Jemné mapování hraničních hodnot u dvou nepříbuzných pacientů se vzácnými překrývajícími se intersticiálními delecemi 9q s mírnými dysmorfickými rysy". American Journal of Medical Genetics. 146A (17): 2234–41. doi:10,1002 / ajmg.a.32397. PMID 18666229.
- ^ Nowakowska B; Stankiewicz P; Obersztyn E; Ou Z; Li J; Chinault AC; Smyk M; Borg K; Mazurczak T; Cheung SW; Bocian E (září 2008). „Aplikace metafáze HR-CGH a cílené chromosomální mikroarray analýzy k genomické charakterizaci 116 pacientů s mentální retardací a dysmorfickými rysy“. American Journal of Medical Genetics. 146A (18): 2361–9. doi:10,1002 / ajmg.a.32475. PMID 18698622.
- ^ Probst FJ; Roeder ER; Enciso VB; Ou Z; Cooper ML; Eng P; Li J; Gu Y; Stratton RF; Chinault AC; Shaw CA; Sutton VR; Cheung SW; Nelson DL (červen 2007). „Chromozomální mikročipová analýza (CMA) detekuje velkou deleci chromozomu X včetně FMR1, FMR2 a IDS u pacientky s mentální retardací“. American Journal of Medical Genetics. 143A (12): 1358–65. doi:10,1002 / ajmg.a.31781. PMID 17506108.
- ^ Hildebrandt, F; et al. (Leden 2009). „Systematický přístup k mapování genů recesivní choroby u jedinců z outbredních populací“. PLOS Genet. 5 (1): e1000353. doi:10.1371 / journal.pgen.1000353. PMC 2621355. PMID 19165332.
- ^ McQuillan R; Leutenegger AL; Abdel-Rahman R; Franklin CS; Pericic M; Barac-Lauc L; Smolej-Narancic N; Janicijevic B; Polasek O; Tenesa A; Macleod AK; Farrington SM; Rudan P; Hayward C; Vitart V; Rudan I; Wild SH; Dunlop MG; Wright AF; Campbell H; Wilson JF (2008). „Běhy homozygotnosti v evropských populacích“. Jsem J Hum Genet. 83 (3): 359–72. doi:10.1016 / j.ajhg.2008.08.007. PMC 2556426. PMID 18760389.
- ^ Gondek LP, Tiu R, O'Keefe CL, Sekeres MA, Theil KS, Maciejewski J (únor 2008). „Chromozomální léze a uniparentální disomie detekované poli SNP v MDS, MDS / MPD a MDS odvozených od AML“. Krev. 111 (3): 1534–42. doi:10.1182 / krev-2007-05-092304. PMC 2214746. PMID 17954704.
- ^ Beroukhim R; Lin M; Park Y; Ha správně; Zhao X; Garraway LA; Fox EA; Hochberg EP; Mellinghoff IK; Hofer MD; Descazeaud A; Rubin MA; Meyerson M; Wong WH; Prodejci WR; Li C (květen 2006). „Odvození ztráty heterozygotnosti z nepárových nádorů pomocí oligonukleotidových polí SNP s vysokou hustotou“. PLOS Comput. Biol. 2 (5): e41. doi:10.1371 / journal.pcbi.0020041. PMC 1458964. PMID 16699594.
- ^ Ishikawa S; Komura D; Tsuji S; Nishimura K; Yamamoto S; Panda B; Huang J; Fukayama M; Jones KW; Aburatani H (srpen 2005). "Analýza alelického dávkování s genotypovacími mikročipy". Biochem Biophys Res Commun. 333 (4): 1309–14. doi:10.1016 / j.bbrc.2005.06.040. PMID 15982637.
- ^ A b Lo KC, Bailey D, Burkhardt T, Gardina P, Turpaz Y, Cowell J (březen 2008). „Komplexní analýza ztráty událostí heterozygotnosti v glioblastomu pomocí mapovacích polí 100K SNP a srovnání s abnormalitami počtu kopií definovanými komparativní genomickou hybridizací pole BAC“. Geny Chromozomy Rakovina. 47 (3): 221–37. doi:10,1002 / gcc.20524. PMID 18050302.
- ^ Mao X, Young BD, Lu Y (červen 2007). „Aplikace mikropolí s jedním nukleotidovým polymorfismem ve výzkumu rakoviny“. Curr Genomics. 8 (4): 219–28. doi:10.2174/138920207781386924. PMC 2430687. PMID 18645599.
- ^ Janoueix-Lerosey I, Schleiermacher G, Michels E a kol. (Březen 2009). „Celkový genomický vzorec je prediktorem výsledku v neuroblastomu“. J. Clin. Oncol. 27 (7): 1026–33. doi:10.1200 / JCO.2008.16.0630. PMID 19171713.
- ^ Michels E, Vandesompele J, Hoebeeck J, Menten B, De Preter K, Laureys G, Van Roy N, Speleman F (2006). „Měření změny počtu kopií DNA v neuroblastomu v celém genomu: disekční amplikony a mapování ztrát, zisků a hraničních hodnot“. Cytogenet. Genome Res. 115 (3–4): 273–282. doi:10.1159/000095924. PMID 17124410.
- ^ Messahel B; Williams R; Ridolfi A; A'hern R; Warren W; Tinworth L; Hobson R; Al-Saadi R; Whyman G; Brundler MA; Kelsey A; Sebire N; Jones C; Vujanic G; Pritchard-Jones K; Dětská skupina pro rakovinu a leukémii (CCLG) (březen 2009). „Dětská skupina pro rakovinu a leukémii (CCLG). Ztráta alely v 16q definuje horší prognózu Wilmsova nádoru bez ohledu na přístup léčby v klinických studiích UKW1-3: studie dětské rakoviny a leukémie (CCLG).“ Eur J Cancer. 45 (5): 819–26. doi:10.1016 / j.ejca.2009.01.005. PMID 19231157.
- ^ Zelený PE; Breslow NE; Li S; Perlman E; Beckwith JB; Ritchey ML; Shamberger RC; Haase GM; D'Angio GJ; Donaldson M; Coppes MJ; Malogolowkin M; Shearer P; Thomas PR; Macklis R; Tomlinson G; Huff V; Green DM; National Wilms Tumor Study Group (October 2005). "National Wilms Tumor Study Group. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group". J Clin Oncol. 23 (29): 7312–21. doi:10.1200/JCO.2005.01.2799. PMID 16129848.
- ^ van den Berg, E; Störkel, S (2003). "Kidney: Clear cell renal cell carcinoma". Atlas Genet Cytogenet Oncol Haematol. 7 (3): 424–431. Citováno 14. prosince 2010.
- ^ Lyons-Weiler MA, Hagenkord JM, Sciulli CM, Dhir R, Monzon F (2008). "Optimization of the Affymetrix GeneChip Mapping 10K 2.0 Assay for Routine Clinical Use on Formalin Fixed Paraffin Embedded Tissues". Diag Mol Path. 17 (1): 3–13. doi:10.1097/PDM.0b013e31815aca30. PMID 18303412.
- ^ Klatte T; Pantuck AJ; Said JW; Seligson DB; Rao NP; LaRochelle JC; Shuch B; Zisman A; Kabbinavar FF; Belldegrun AS (2009). "Cytogenetic and molecular tumor profiling for type 1 and type 2 papillary renal cell carcinoma". Klinický výzkum rakoviny. 15 (4): 1162–9. doi:10.1158/1078-0432.CCR-08-1229. PMID 19228721.
- ^ Brunelli M; Eccher A; Gobbo S; Ficarra V; Novara G; Cossu-Rocca P; Bonetti F; Menestrina F; Cheng L; Eble JN; Martignoni G (2008). "Loss of chromosome 9p is an independent prognostic factor in patients with clear cell renal cell carcinoma". Moderní patologie. 21 (1): 1–6. doi:10.1038/modpathol.3800967. PMID 17906617.
- ^ Klatte T; Rao PN; de Martino M; LaRochelle J; Shuch B; Zomorodian N; Said J; Kabbinavar FF; Belldegrun AS; Pantuck AJ (2009). "Cytogenetic profile predicts prognosis of patients with clear cell renal cell carcinoma". Journal of Clinical Oncology. 27 (5): 746–53. doi:10.1200/JCO.2007.15.8345. PMID 19124809.
- ^ Szponar A, Zubakov D, Pawlak J, Jauch A, Kovacs G (2009). "Three genetic developmental stages of papillary renal cell tumors: duplication of chromosome 1q marks fatal progression". International Journal of Cancer. 124 (9): 2071–6. doi:10.1002/ijc.24180. PMID 19123481.
- ^ Schwaenen C; Nessling M; Wessendorf S; Salvi T; Wrobel G; Radlwimmer B; Kestler HA; Haslinger C; Stilgenbauer S; Döhner H; Bentz M; Lichter P (2004). "Automated array-based genomic profiling in chronic lymphocytic leukemia: development of a clinical tool and discovery of recurrent genomic alterations". Proc Natl Acad Sci U S A. 101 (4): 1039–44. doi:10.1073/pnas.0304717101. PMC 327147. PMID 14730057.
- ^ Pfeifer D; Pantic M; Skatulla I; Rawluk J; Kreutz C; Martens UM; Fisch P; Timmer J; Veelken H (February 2007). "Genome-wide analysis of DNA copy number changes and LOH in CLL using high-density SNP arrays". Krev. 109 (3): 1202–10. doi:10.1182/blood-2006-07-034256. PMID 17053054.
- ^ Gunn SR; Mohammed MS; Gorre ME; Cotter PD; Kim J; Bahler DW; Preobrazhensky SN; Higgins RA; Bolla AR; Ismail SH; de Jong D; Eldering E; van Oers MH; Mellink CH; Keating MJ; Schlette EJ; Abruzzo LV; Robetorye RS (September 2008). "Whole-genome scanning by array comparative genomic hybridization as a clinical tool for risk assessment in chronic lymphocytic leukemia". The Journal of Molecular Diagnostics. 10 (5): 442–451. doi:10.2353/jmoldx.2008.080033. PMC 2518739. PMID 18687794.
- ^ Sargent R; Jones D; Abruzzo LV; Yao H; Bonderover J; Cisneros M; Wierda WG; Keating MJ; Luthra R (January 2009). "Customized oligonucleotide array-based comparative genomic hybridization as a clinical assay for genomic profiling of chronic lymphocytic leukemia". J Mol Diagn. 11 (1): 25–34. doi:10.2353/jmoldx.2009.080037. PMC 2607562. PMID 19074592.
- ^ 2009 May;23(5):829-33
- ^ Hagenkord JM, Monzon FA, Kash SF, Lilleberg S, Xie Q, Kant JA (2010). "Array-based karyotyping for prognostic assessment in chronic lymphocytic leukemia: performance comparison of affymetrix 10K2.0, 250K Nsp, and SNP6.0 arrays". J Mol Diagn. 12 (2): 184–96. doi:10.2353/jmoldx.2010.090118. PMC 2871725. PMID 20075210.
- ^ Dohner H, Stilgenbauer S, Benner A, et al. (2000). "Genomic aberrations and survival in chronic lymphocytic leukemia". NEJM. 343 (26): 1910–6. doi:10.1056 / NEJM200012283432602. PMID 11136261.
- ^ Hervé Avet-Loiseau; Cheng Li; Florence Magrangeas; Wilfried Gouraud; Catherine Charbonnel; Jean-Luc Harousseau; Michel Attal; Gerald Marit; Claire Mathiot; Thierry Facon; Philippe Moreau; Kenneth C. Anderson; Loïc Campion; Nikhil C. Munshi; Stéphane Minvielle (September 2009). "Prognostic significance of copy-number alterations in multiple myeloma". Journal of Clinical Oncology. 27 (27): 4585–90. doi:10.1200/JCO.2008.20.6136. PMC 2754906. PMID 19687334.
- ^ Pfister S; Remke M; Benner A; Mendrzyk F; Toedt G; Felsberg J; Wittmann A; Devens F; Gerber NU; Joos S; Kulozik A; Reifenberger G; Rutkowski S; Wiestler OD; Radlwimmer B; Scheurlen W; Lichter P; Korshunov A (April 2009). "Outcome Prediction in Pediatric Medulloblastoma based on DNA Copy Number Aberrations of Chromosomes 6q and 17q and the MYC and MYCN Loci". J Clin Oncol. 27 (10): 1627–1636. doi:10.1200 / JCO.2008.17.9432. PMID 19255330.
- ^ A b Barbashina V, Salazar P, Holland EC, Rosenblum MK, Ladanyi M (1 February 2005). "Allelic losses at 1p36 and 19q13 in gliomas: correlation with histologic classification, definition of a 150-kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene". Clin. Cancer Res. 11 (3): 1119–28. PMID 15709179.
- ^ Laigle-Donadey F, Benouaich-Amiel A, Hoang-Xuan K, Sanson M (2005). "[Molecular biology of oligodendroglial tumors]". Neuro-Chirurgie (francouzsky). 51 (3–4 Pt 2): 260–8. doi:10.1016/s0028-3770(05)83487-3. PMID 16292170.
- ^ Walker C, Haylock B, Husband D, et al. (2006). "Clinical use of genotype to predict chemosensitivity in oligodendroglial tumors". Neurologie. 66 (11): 1661–7. doi:10.1212/01.wnl.0000218270.12495.9a. PMID 16769937.
- ^ Jenkins RB, Blair H, Ballman KV, et al. (Říjen 2006). "A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma". Cancer Res. 66 (20): 9852–61. doi:10.1158/0008-5472.CAN-06-1796. PMID 17047046.
- ^ Ohgaki H, Eibl RH, Wiestler OD, Yasargil MG, Newcomb EW, Kleihues P (15 November 1991). "mutace p53 v nonastrocytárních lidských mozkových nádorech". Cancer Res. 51 (22): 6202–5. PMID 1933879.
- ^ Ducray F, Idbaih A, de Reyniès A, et al. (2008). "Anaplastic oligodendrogliomas with 1p19q codeletion have a proneural gene expression profile". Mol. Rakovina. 7 (1): 41. doi:10.1186/1476-4598-7-41. PMC 2415112. PMID 18492260.
- ^ Dong Yin; Seishi Ogawa; Norihiko Kawamata; Patrizia Tunici; Gaetano Finocchiaro; Marica Eoli; Christian Ruckert; Thien Huynh; Gentao Liu; Motohiro Kato; Masashi Sanada; Anna Jauch; Martin Dugas; Keith L. Black; H. Phillip Koeffler (May 2009). "High-Resolution Genomic Copy Number Profiling of Glioblastoma Multiforme by Single Nucleotide Polymorphism DNA Microarray". Mol Cancer Res. 7 (5): 665–77. doi:10.1158/1541-7786.MCR-08-0270. PMID 19435819.
- ^ Cancer Cytogenetics, 3rd Ed, Chapter 19, Tumors of the Nervous System, Wiley Blackwell 2009.
- ^ Tumors of the Central Nervous System. Vol 7. Washington DC: American Registry of Pathology; 2007
- ^ Moorman A, Harrison C, Buck G, Richards S, Secker-Walker L, Martineau M, Vance G, Cherry A, Higgins R, Fielding A, Foroni L, Paietta E, Tallman M, Litzow M, Wiernik P, Rowe J, Goldstone A, Dewald G (2007). „Karyotyp je nezávislým prognostickým faktorem u dospělé akutní lymfoblastické leukémie (ALL): analýza cytogenetických údajů od pacientů léčených ve studii Medical Research Council (MRC) UKALLXII / Eastern Cooperative Oncology Group (ECOG) 2993“. Krev. 109 (8): 3189–97. doi:10.1182 / krev-2006-10-051912. PMID 17170120.
- ^ Kawamata N; Ogawa S; Zimmermann M; Kato M; Sanada M; Hemminki K; Yamatomo G; Nannya Y; Koehler R; Flohr T; Miller CW; Harbott J; Ludwig WD; Stanulla M; Schrappe M; Bartram CR; Koeffler HP (January 2008). "Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high-resolution single nucleotide polymorphism oligonucleotide genomic microarray". Krev. 111 (2): 776–84. doi:10.1182/blood-2007-05-088310. PMC 2200831. PMID 17890455.
- ^ Bungaro S; Dell'Orto MC; Zangrando A; Basso D; Gorletta T; Lo Nigro L; Leszl A; Young BD; Basso G; Bicciato S; Biondi A; te Kronnie G; Cazzaniga G (January 2009). "Integration of genomic and gene expression data of childhood ALL without known aberrations identifies subgroups with specific genetic hallmarks". Geny Chromozomy Rakovina. 48 (1): 22–38. doi:10.1002/gcc.20616. PMID 18803328.
- ^ Sulong S; Moorman AV; Irving JA; Strefford JC; Konn ZJ; Case MC; Minto L; Barber KE; Parker H; Wright SL; Stewart AR; Bailey S; Bown NP; Hall AG; Harrison CJ (January 2009). "A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups". Krev. 113 (1): 100–7. doi:10.1182/blood-2008-07-166801. PMID 18838613.
- ^ Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. (Leden 2009). „Podtyp dětské akutní lymfoblastické leukémie se špatným výsledkem léčby: klasifikační studie pro celý genom“. Lancet Oncol. 10 (2): 125–34. doi:10.1016 / S1470-2045 (08) 70339-5. PMC 2707020. PMID 19138562.
- ^ Hasse D (2008). "Cytogenetic features in myelodysplastic syndromes". Ann Hematol. 87 (7): 515–526. doi:10.1007/s00277-008-0483-y. PMC 2413090. PMID 18414863.
- ^ WHO Classification of Tumours of Haematopoeitic and Lymphoid Tissues, Edited by Swerdlow SH, et al. IARC Press, 2008, Lyon.
- ^ Makishima H; Rataul M; Gondek LP; Huh J; Cook JR; Theil KS; Sekeres MA; Kuczkowski E; O'Keefe C; Maciejewski JP (2010). "FISH and SNP-A karyotyping in myelodysplastic syndromes: Improving cytogenetic detection of del(5q), monosomy 7, del(7q), trisomy 8, and del(20q)". Leuk Res. 34 (4): 447–453. doi:10.1016/j.leukres.2009.08.023. PMC 2826525. PMID 19758696.
- ^ Sanada, et al. "Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms." Příroda 13 Aug 2009; 460, 904–909.
- ^ Gondek LP, Tiu R, O'Keefe CL, Sekeres MA, Theil KS, MacIejewski JP (2008). "Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML". Krev. 111 (3): 1534–42. doi:10.1182/blood-2007-05-092304. PMC 2214746. PMID 17954704.
- ^ Thoennissen NH; Krug UO; Lee DH; Kawamata N; Iwanski GB; Lasho T; Weiss T; Nowak D; Koren-Michowitz M; Kato M; Sanada M; Shih LY; Nagler A; Raynaud SD; Müller-Tidow C; Mesa R; Haferlach T; Gilliland DG; Tefferi A; Ogawa S; Koeffler HP (April 2010). "Prevalence and prognostic impact of allelic imbalances associated with leukemic transformation of Philadelphia chromosome-negative myeloproliferative neoplasms". Krev. 115 (14): 2882–2890. doi:10.1182/blood-2009-07-235119. PMC 2854432. PMID 20068225.
- ^ A b Lenz HJ, "Established Biomarkers for Colorectal Carcinoma", American Society of Clinical Oncology Educational Book, 2009, p215-219.
- ^ A b C Jackson EM; Sievert AJ; Gai X; Hakonarson H; Judkins AR; Tooke L; Perin JC; Xie H; Shaikh TH; Biegel JA (2009). "Genomic analysis using high density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides comprehensive analysis of INI1/SMARCB1 in Malignant Rhabdoid Tumors". Clin Cancer Res. 15 (6): 1923–1930. doi:10.1158 / 1078-0432.CCR-08-2091. PMC 2668138. PMID 19276269.
- ^ Prescher G, Bornfeld N, Hirche H, Horsthemke B, Jöckel KH, Becher R (1996). "Prognostic implications of monosomy 3 in uveal melanoma". Lanceta. 347 (9010): 1222–1225. doi:10.1016/S0140-6736(96)90736-9. PMID 8622452.
- ^ Damato BE, Dopierala J, Klaasen A, van Dijk M, Sibbring J, Coupland S (2009). "Multiplex Ligation-Dependent Probe Amplification of Uveal Melanoma: Correlation with Metastatic Death" (PDF). Investujte Ophthalmol Vis Sci. 50 (7): 3048–55. doi:10.1167/iovs.08-3165. PMID 19182252.
- ^ White VA, McNeil BK, Horsman DE (1998). "Acquired homozygosity (isodisomy) of chromosome 3 in uveal melanoma". Cancer Genet Cytogenet. 102 (1): 40–45. doi:10.1016/S0165-4608(97)00290-2. PMID 9530338.
- ^ Onken MD, Worley LA, Person E, Char DH, Bowcock AM, Harbour JW (2007). "Loss of heterozygosity of chromosome 3 detected with single nucleotide polymorphisms is superior to monosomy 3 for predicting metastasis in uveal melanoma". Clin Cancer Res. 13 (10): 2923–2937. doi:10.1158/1078-0432.CCR-06-2383. PMID 17504992.
- ^ Yamamoto G; Nannya Y; Kato M; Sanada M; Levine RL; Kawamata N; Hangaishi A; Kurokawa M; Chiba S; Gilliland DG; Koeffler HP; Ogawa S (July 2007). "Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of affymetrix single-nucleotide-polymorphism genotyping microarrays". Jsem J Hum Genet. 81 (1): 114–26. doi:10.1086/518809. PMC 1950910. PMID 17564968.