K-mer - K-mer

v bioinformatika, k-merové jsou podsekvence délky obsažené v biologické sekvenci. Primárně se používá v kontextu výpočetní genomika a sekvenční analýza, ve kterém k-merové se skládají z nukleotidy (tj. A, T, G a C), k-merové jsou velkými písmeny sestavte sekvence DNA,[1] zlepšit heterologní genová exprese,[2][3] identifikovat druhy v metagenomických vzorcích,[4] a tvořit oslabené vakcíny.[5] Obvykle termín k-mer označuje všechny subsekvence sekvence délky , takže sekvence AGAT bude mít čtyři monomery (A, G, A a T), tři 2-mer (AG, GA, AT), dva 3-mer (AGA a GAT) a jeden 4-mer (AGAT). Obecněji posloupnost délky budu mít k-majitelé a celkem možné k-meri, kde je počet možných monomerů (např. čtyři v případě DNA ).





Úvod
k-merové jsou prostě délka podsekvence. Například vše možné k- polymery sekvence DNA jsou uvedeny níže:
| k | k-merové |
|---|---|
| 1 | G, T, A, G, A, G, C, T, G, T |
| 2 | GT, TA, AG, GA, AG, GC, CT, TG, GT |
| 3 | GTA, TAG, AGA, GAG, AGC, GCT, CTG, TGT |
| 4 | GTAG, TAGA, AGAG, GAGC, AGCT, GCTG, CTGT |
| 5 | GTAGA, TAGAG, AGAGC, GAGCT, AGCTG, GCTGT |
| 6 | GTAGAG, TAGAGC, AGAGCT, GAGCTG, AGCTGT |
| 7 | GTAGAGC, TAGAGCT, AGAGCTG, GAGCTGT |
| 8 | GTAGAGCT, TAGAGCTG, AGAGCTGT |
| 9 | GTAGAGCTG, TAGAGCTGT |
| 10 | GTAGAGCTGT |
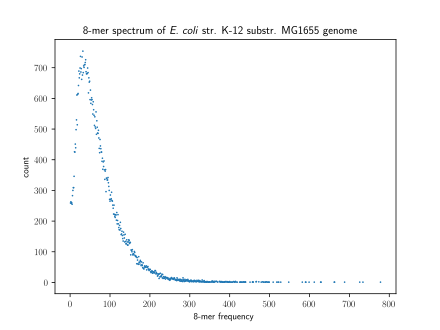
Způsob vizualizace k-merové, k-mer spektrum, ukazuje multiplicitu každého z nich k-mer v pořadí versus počet k-majitelé s touto multiplicitou.[6] Počet režimů v a k-merní spektrum pro genom druhu se liší, přičemž většina druhů má unimodální distribuci.[7] Však všichni savci mít multimodální distribuci. Počet režimů v rámci a k-merní spektrum se může lišit i mezi oblastmi genomu: lidé mají unimodální k-mer spektra dovnitř 5 'UTR a exony ale multimodální spektra v 3 'UTR a introny.
Síly ovlivňující DNA k-mer frekvence
Četnost k-používání uživatelů je ovlivňováno četnými silami, pracujícími na více úrovních, které jsou často v konfliktu. Je důležité si to uvědomit k-mery pro vyšší hodnoty k jsou ovlivněny silami ovlivňujícími nižší hodnoty k také. Pokud se například 1-mer A nevyskytuje v sekvenci, nedojde ani k jednomu ze 2-merů obsahujících A (AA, AT, AG a AC), čímž dojde k propojení účinků různých sil.
k = 1
Když k = 1, existují čtyři DNA k-merové, tj.„A, T, G a C. Na molekulární úrovni existují tři Vodíkové vazby mezi G a C, zatímco mezi A a T jsou pouze dvě. GC vazby jsou v důsledku extra vodíkové vazby (a silnějších interakcí stohování) tepelně stabilnější než AT vazby.[8] Savci a ptáci mají vyšší poměr Gs a Cs k As a Ts (Obsah GC ), což vedlo k hypotéze, že tepelná stabilita byla hybným faktorem variace obsahu GC.[9] Tato hypotéza, i když byla slibná, neobstála pod kontrolou: analýza mezi různými prokaryoty neprokázala žádný důkaz obsahu GC v korelaci s teplotou, jak by předpovídala hypotéza tepelné adaptace.[10] Pokud by hnací silou za variací obsahu GC měl být přirozený výběr, to by to vyžadovalo změny jednoho nukleotidu, které jsou často tichý, změnit kondici organismu.[11]
Současné důkazy to spíše naznačují Konverze genu ovlivněná GC (gBGC) je hnacím faktorem variací obsahu GC.[11] gBGC je proces, ke kterému dochází během rekombinace který nahradí Gs a Cs As a Ts.[12] Tento proces, i když se liší od přirozeného výběru, může přesto vyvinout selektivní tlak na DNA předpjatou směrem k nahrazení GC fixovaným v genomu. gBGC lze proto považovat za „podvodníka“ přirozeného výběru. Jak by se dalo očekávat, obsah GC je vyšší na webech, kde dochází k větší rekombinaci.[13] Kromě toho organismy s vyšší rychlostí rekombinace vykazují vyšší obsah GC v souladu s předpokládanými účinky hypotézy gBGC.[14] Zajímavé je, že se zdá, že gBGC není omezen na eukaryoty.[15] Nepohlavní organismy, jako jsou bakterie a archea, také zažívají rekombinaci pomocí genové konverze, což je proces nahrazení homologní sekvence, jehož výsledkem je více identických sekvencí v celém genomu.[16] Že je rekombinace schopna zvýšit obsah GC ve všech doménách života, naznačuje, že gBGC je všeobecně konzervovaný. Je třeba určit, zda je gBGC (většinou) neutrálním vedlejším produktem molekulárního aparátu života nebo zda je sám pod selekcí. Přesný mechanismus a evoluční výhody nebo nevýhody gBGC nejsou v současné době známy.[17]
k = 2
Navzdory poměrně velkému množství literatury pojednávající o předsudcích obsahujících GC, bylo o předsudcích dinukleotidů napsáno relativně málo. Je známo, že tyto předsudky dinukleotidů jsou relativně konstantní v celém genomu, na rozdíl od obsahu GC, který, jak je vidět výše, se může značně lišit.[18] Jedná se o důležitý pohled, který nelze přehlédnout. Pokud byly zkreslení dinukleotidů vystaveny tlakům vyplývajícím z překlad, pak by existovaly různé vzorce zkreslení dinukleotidů kódování a nekódující regiony poháněné sníženou translační účinností některých dinucelotidů.[19] Protože tomu tak není, lze proto odvodit, že síly modulující zkreslení dinukleotidů jsou nezávislé na translaci. Dalším důkazem proti translačním tlakům ovlivňujícím předpětí dinukleotidů je skutečnost, že předpětí dinukleotidů virů, které se do značné míry spoléhají na translační účinnost, jsou více formovány jejich virovou rodinou než hostiteli, jejichž translační aparát viry unesou.[20]
Proti rostoucímu obsahu GC v gBGC je Potlačení CG, což snižuje frekvenci CG 2 měsíce kvůli deaminace z methylovaný CG dinukleotidy, což vede k substituci CG za TG, čímž se snižuje obsah GC.[21] Tato interakce zdůrazňuje vzájemný vztah mezi působícími silami k-mers pro různé hodnoty k.
Jedním zajímavým faktem o zaujatosti dinukleotidů je, že může sloužit jako měření „vzdálenosti“ mezi fylogeneticky podobnými genomy. Genomy párů organismů, které spolu úzce souvisejí, sdílejí podobnější předsudky dinukleotidů než mezi páry vzdáleněji příbuzných organismů.[18]
k = 3
Existuje dvacet přirozených aminokyseliny které se používají k vytváření proteinů, které DNA kóduje. Existují však pouze čtyři nukleotidy. Proto nemůže existovat vzájemná korespondence mezi nukleotidy a aminokyselinami. Podobně existuje 16 2-merů, což také nestačí k tomu, aby jednoznačně představovalo každou aminokyselinu. V DNA však existuje 64 odlišných 3-merů, což je dostatečné množství pro jedinečnou reprezentaci každé aminokyseliny. Tito nepřekrývající se 3-mery se nazývají kodony. Zatímco každý kodon mapuje pouze jednu aminokyselinu, každá aminokyselina může být reprezentované několika kodony. Stejná aminokyselinová sekvence tedy může mít více reprezentací DNA. Je zajímavé, že každý kodon pro aminokyselinu není použit ve stejném poměru.[22] Tomu se říká zkreslení použití kodonů (MLÁDĚ). Když k = 3, je třeba rozlišovat mezi skutečnou 3mernou frekvencí a CUB. Například sekvence ATGGCA má v sobě čtyři 3-merní slova (ATG, TGG, GGC a GCA), přičemž obsahuje pouze dva kodony (ATG a GCA). CUB je však hlavním hnacím faktorem zkreslení používání 3 merů (tvoří až ⅓ toho, protože ⅓ k- výrobci v kódující oblasti jsou kodony) a budou hlavním zaměřením této části.
Přesná příčina variace mezi frekvencemi různých kodonů není zcela objasněna. Je známo, že preference kodonů koreluje s výskytem tRNA, přičemž kodony odpovídající hojnějším tRNA jsou odpovídajícím způsobem častější[22] a že více vysoce exprimované proteiny vykazují vyšší CUB.[23] To naznačuje, že výběr pro translační účinnost nebo přesnost je hnací silou variace CUB.
k = 4
Podobně jako účinek pozorovaný u předpětí dinukleotidů, předpětí tetranukleotidů u fylogeneticky podobných organismů jsou podobnější než u méně blízkých organismů.[4] Přesná příčina variace předpětí tetranukleotidů není dobře známa, ale předpokládá se, že je výsledkem udržení genetické stability na molekulární úrovni.[24]
Aplikace
Frekvence sady kjako "podpis" podkladové sekvence lze použít polymery v genomu druhu, v genomové oblasti nebo ve třídě sekvencí. Porovnání těchto frekvencí je výpočetně snazší než zarovnání sekvence, a je důležitou metodou v sekvenční analýza bez zarovnání. Lze jej také použít jako analýzu prvního stupně před zarovnáním.
Sekvenční sestava

V sestavě sekvence, k-mery se používají při stavbě De Bruijn grafy.[25][26] Aby bylo možné vytvořit graf De Bruijn, k-mery uložené v každé hraně s délkou musí překrývat další řetězec na jiném okraji o za účelem vytvoření vrchol. Čtení generovaná z sekvenování nové generace bude mít obvykle generované různé délky čtení. Například čte podle Illumina Sekvenční technologie zachycuje čtení 100 metrů. Problém se sekvenováním však je, že ve skutečnosti jsou generovány pouze malé zlomky ze všech možných 100-merů, které jsou přítomny v genomu. To je způsobeno chybami čtení, ale co je důležitější, jen jednoduché díry pokrytí, které se vyskytnou během sekvenování. Problém je v tom, že tyto malé zlomky možných k-merové porušují klíčový předpoklad grafů De Bruijna, že všechny k-mer čtení musí překrývat jeho sousední k-mír v genomu (což nemůže nastat, když je to možné k-merové nejsou přítomni).


Řešením tohoto problému je prolomit je k- menší velikost čte na menší k-mers, tak, že výsledný menší k-merové budou představovat vše možné k-majitelé této menší velikosti, kteří jsou přítomni v genomu.[27] Dále rozdělení k-mers do menších velikostí také pomáhá zmírnit problém různých počátečních délek čtení. V tomto příkladu pět čtení nezohledňuje všechny možné 7mery genomu a jako takový nelze vytvořit graf De Bruijn. Ale když jsou rozděleny na 4-mery, výsledné subsekvence stačí k rekonstrukci genomu pomocí De Bruijnova grafu.
Kromě přímého použití pro sestavení sekvence, kLátky lze také použít k detekci nesprávného sestavení genomu identifikací k- výrobci, kteří jsou nadměrně zastoupeni, což naznačuje přítomnost opakované sekvence DNA které byly kombinovány.[28] Navíc, k-mery se také používají k detekci bakteriální kontaminace během shromáždění eukaryotického genomu, což je přístup převzatý z oblasti metagenomiky.[29][30]
Volba k-mer
Volba k-mer size has many different effects on the sequence assembly. Tyto efekty se velmi liší mezi menšími a většími k-merové. Proto pochopení různých k-Musí být dosaženo velikosti, aby bylo možné zvolit vhodnou velikost, která vyvažuje efekty. Účinky velikostí jsou uvedeny níže.
Dolní k-merní velikosti
- Nižší k-mer velikost sníží množství okrajů uložených v grafu a jako takové pomůže snížit množství prostoru potřebného pro uložení sekvence DNA.
- Mít menší velikosti zvýší šanci pro všechny k-mers to overlap, and as such, have the required subsekces in order to construct the De Bruijn graph.[31]
- Tím, že má menší velikost k-meri, také riskujete, že budete mít v grafu mnoho vrcholů vedoucích do jednoho k-meru. To proto zkomplikuje rekonstrukci genomu, protože existuje vyšší úroveň nejednoznačností dráhy kvůli většímu počtu vrcholů, které bude třeba projít.
- Informace jsou ztraceny jako k-merové se zmenšují.
- Např. Možnost AGTCGTAGATGCTG je nižší než ACGT a jako taková obsahuje větší množství informací (viz entropie (teorie informace) Pro více informací).
- Menší kVýrobci mají také problém, že nejsou schopni vyřešit oblasti v DNA, kde jsou malé mikrosatelity nebo se opakují. Je to proto, že menší k-majitelé budou mít tendenci sedět úplně v oblasti opakování, a proto je těžké určit míru opakování, ke které skutečně došlo.
- Např. U subsekvence ATGTGTGTGTGTGTACG bude množství opakování TG ztraceno, pokud k- je vybrána velikost menší než 16. Je to proto, že většina z k-merové budou sedět v opakované oblasti a mohou být zahozeni jako jejich opakování k-mer místo odkazování na množství opakování.
Vyšší k-merní velikosti
- S větší velikostí k-mery zvýší počet hran v grafu, což zase zvýší množství paměti potřebné k uložení sekvence DNA.
- Zvětšením velikosti k-merů, sníží se také počet vrcholů. To pomůže s konstrukcí genomu, protože v grafu bude méně cest k procházení.[31]
- Větší k-merové také riskují, že nebudou mít vnější vrcholy z každého k-meru. To je způsobeno větším k- výrobci zvyšující riziko, že se nebudou překrývat s jinými k-mer by . To proto může vést k nesouvislosti ve čtení, a jako takové, může vést k vyššímu množství menších kontigy.
- Větší k-merní velikosti pomáhají zmírnit problém malých regionů s opakováním. To je způsobeno skutečností, že k-mer bude obsahovat rovnováhu oblasti opakování a sousedních sekvencí DNA (vzhledem k tomu, že jsou dostatečně velké velikosti), které mohou pomoci vyřešit množství opakování v dané konkrétní oblasti.
Genetika a genomika
Pokud jde o onemocnění, bylo k detekci genetických ostrovů spojených s patogenitou aplikováno zkreslení dinukleotidy.[11] Předchozí práce také ukázaly, že předpětí tetranukleotidů jsou schopny účinně detekovat horizontální přenos genů u obou prokaryot[32] a eukaryoty.[33]
Další aplikace k-mers je v taxonomii založené na genomice. Například obsah GC byl použit k rozlišení mezi druhy Erwinia s mírným úspěchem.[34] Podobné přímému použití obsahu GC pro taxonomické účely je použití Tm, teplota tání DNA. Protože vazby GC jsou tepelně stabilnější, sekvence s vyšším obsahem GC vykazují vyšší Tm. V roce 1987 Výbor ad hoc pro usmíření přístupů k bakteriální systematice navrhl použití ΔTm jako faktor při určování hranic druhů jako součást koncept fylogenetických druhů, ačkoli se nezdá, že by tento návrh získal vědeckou komunitu.[35]
Mezi další aplikace v genetice a genomice patří:
- RNA izoforma kvantifikace od RNA sekvence data[36]
- Klasifikace lidských mitochondrií haploskupina[37]
- Detekce rekombinačních míst v genomech[38]
- Odhad velikost genomu použitím k-mer frekvence vs. k-mer hloubka[39][40]
- Charakterizace CpG ostrovy sousedními regiony[41][42]
- De novo detekce opakovaná sekvence jako transponovatelný prvek[43]
- Čárový kód DNA druhů.[7][44]
- Charakterizace vazby na bílkoviny sekvenční motivy[45]
- Identifikace mutace nebo polymorfismus pomocí nové generace sekvenování data[46]
Metagenomika
k-merová frekvence a variace spektra se v metagenomice pro obě analýzy silně používají[47][48] a binování. Při binningu je úkolem oddělit sekvenční čtení do „košů“ čtení pro každý organismus (nebo funkční taxonomická jednotka ), které budou poté smontovány. TETRA je pozoruhodný nástroj, který odebírá metagenomické vzorky a ukládá je do organismů na základě jejich tetranukleotidů (k = 4) frekvence.[49] Další nástroje, na které se podobně spoléhají k-mer frekvence pro metagenomické binování jsou CompostBin (k = 6),[50] PCAHIER,[51] PhyloPythia (5 ≤ k ≤ 6),[52] CLARK (k ≥ 20),[53] a TACOA (2 ≤k ≤ 6).[54] Rovněž se uplatnil nejnovější vývoj hluboké učení na metagenomické binování pomocí k-merové.[55]
Mezi další aplikace v rámci metagenomiky patří:
- Obnova čtecích rámců ze surových čtení[56]
- Odhad početnosti druhů v metagenomických vzorcích[57]
- Určení, které druhy jsou přítomny ve vzorcích[58][59]
- Identifikace biomarkery pro choroby ze vzorků[60]
Biotechnologie
Úpravy k-merní frekvence v sekvencích DNA byly značně používány v biotechnologických aplikacích k řízení translační účinnosti. Konkrétně se používá k regulaci rychlosti produkce proteinu nahoru i dolů.
S ohledem na zvýšení produkce bílkovin bylo použito snížení nepříznivé frekvence dinukleotidů, což vedlo k vyšší rychlosti syntézy bílkovin.[61] Kromě toho byla upravena předpětí využití kodonů, aby se vytvořily synonymní sekvence s vyšší rychlostí exprese proteinu.[2][3] Podobně byla pro zvýšení exprese také úspěšně použita optimalizace kodonových párů, kombinace dinucelotidu a optimalizace kodonů.[62]
Nejvíce studovaná aplikace kpro snížení účinnosti translace je manipulace s kodonovými páry pro zeslabení virů za účelem vytvoření vakcín. Vědci dokázali překódovat virus dengue, virus, který způsobuje horečka dengue, takže jeho předpětí kodonových párů se více lišilo od preferencí použití kodonů savců než u divokého typu.[63] Ačkoli obsahoval identickou aminokyselinovou sekvenci, překódovaný virus prokázal významně oslabený patogenita při vyvolání silné imunitní odpovědi. Tento přístup byl také účinně použit k vytvoření vakcíny proti chřipce[64] stejně vakcína pro Marekova choroba herpesvirus (MDV).[65] Zejména manipulace se zkreslením kodonových párů použitá k oslabení MDV účinně nesnížila onkogenicita viru, což zdůrazňuje potenciální slabost v biotechnologických aplikacích tohoto přístupu. K dnešnímu dni nebyla pro použití schválena žádná deoptimizovaná vakcína s kodonovými páry.
Dva pozdější články pomáhají vysvětlit skutečný mechanismus, který je základem deoptimizace kodonových párů: zkreslení kodonových párů je výsledkem zkreslení dinukleotidů.[66][67] Studiem virů a jejich hostitelů dokázaly obě skupiny autorů dospět k závěru, že molekulárním mechanismem, který vede k pozorování virů, je nárůst dinukleotidů, které jsou špatně vhodné pro překlad.
Obsah GC, kvůli jeho účinku na Teplota tání DNA, se používá k předpovědi teploty žíhání v PCR, další důležitý biotechnologický nástroj.
Implementace
Pseudo kód
Určení možného k-many čtení lze provést jednoduchým cyklováním přes délku řetězce o jednu a vyjmutím každého dílčího řetězce délky . K dosažení tohoto cíle je pseudokód následující:
postup k-mers (řetězec seq, celé číslo k) je L ← délka (seq) arr ← nové pole L - k + 1 prázdné řetězce // iterace nad počtem k-mers v seq, // uložení n-tého k-meru do výstupního pole pro n ← 0 na L - k + 1 exkluzivně dělat arr [n] ← subsekvence sekv. od písmene n včetně po písmeno n + k bez vrátit se přílet
In Bioinformatics Pipelines
Protože počet k-mers roste exponenciálně pro hodnoty k, počítání k-mers pro velké hodnoty k (obvykle> 10) je výpočetně obtížný úkol. Zatímco jednoduché implementace, jako je výše uvedený pseudokód, fungují pro malé hodnoty k, je třeba je přizpůsobit pro aplikace s vysokou propustností nebo kdy k je velký. K vyřešení tohoto problému byly vyvinuty různé nástroje:
- Medúza používá vícevláknové, bezzámkové hash tabulka pro k-mer počítá a má Krajta, Rubín, a Perl vazby[68]
- KMC je nástroj pro k-mer počítání, které používá vícevidovou architekturu pro optimalizovanou rychlost[69]
- Gerbil používá přístup hash tabulky, ale s přidanou podporou akcelerace GPU[70]
- Sada nástrojů pro analýzu K-mer (KAT) používá k analýze upravenou verzi medúzy k-mer se počítá[6]
Viz také
Reference
 Část obsahu v tomto článku byla zkopírována K-mer na wiki systému PLOS, který je k dispozici pod a Obecná licence Creative Commons Attribution 2.5 (CC BY 2.5).
Část obsahu v tomto článku byla zkopírována K-mer na wiki systému PLOS, který je k dispozici pod a Obecná licence Creative Commons Attribution 2.5 (CC BY 2.5).
- ^ Compeau, Phillip E C; Pevzner, Pavel A; Tesler, Glenn (listopad 2011). „Jak aplikovat de Bruijnovy grafy na genomové seskupení“. Přírodní biotechnologie. 29 (11): 987–991. doi:10.1038 / nbt.2023. ISSN 1087-0156. PMC 5531759. PMID 22068540.
- ^ A b Welch, Mark; Govindarajan, Sridhar; Ness, Jon E .; Villalobos, Alan; Gurney, Austin; Minshull, Jeremy; Gustafsson, Claes (2009-09-14). Kudla, Grzegorz (ed.). "Konstrukční parametry pro řízení exprese syntetického genu v Escherichia coli". PLOS ONE. 4 (9): e7002. Bibcode:2009PLoSO ... 4,7002W. doi:10,1371 / journal.pone.0007002. ISSN 1932-6203. PMC 2736378. PMID 19759823.
- ^ A b Gustafsson, Claes; Govindarajan, Sridhar; Minshull, Jeremy (červenec 2004). "Předpětí kodonu a exprese heterologního proteinu". Trendy v biotechnologii. 22 (7): 346–353. doi:10.1016 / j.tibtech.2004.04.006. PMID 15245907.
- ^ A b Perry, Scott C .; Beiko, Robert G. (01.01.2010). „Rozlišování fragmentů mikrobiálních genomů na základě jejich složení: evoluční a srovnávací genomové perspektivy“. Biologie genomu a evoluce. 2: 117–131. doi:10.1093 / gbe / evq004. ISSN 1759-6653. PMC 2839357. PMID 20333228.
- ^ Eschke, Kathrin; Trimpert, Jakob; Osterrieder, Nikolaus; Kunec, Dušan (29.01.2018). Mocarski, Edward (ed.). „Útlum velmi virulentního herpesviru Marekovy choroby (MDV) deoptimizací předpětí kodonových párů“. PLOS patogeny. 14 (1): e1006857. doi:10.1371 / journal.ppat.1006857. ISSN 1553-7374. PMC 5805365. PMID 29377958.
- ^ A b Mapleson, Daniel; Garcia Accinelli, Gonzalo; Kettleborough, George; Wright, Jonathan; Clavijo, Bernardo J. (2016-10-22). „KAT: Sada nástrojů pro analýzu K-mer ke kontrole kvality datových souborů NGS a genomových sestav“. Bioinformatika. 33 (4): 574–576. doi:10.1093 / bioinformatika / btw663. ISSN 1367-4803. PMC 5408915. PMID 27797770.
- ^ A b Chor, Benny; Horn, David; Goldman, Nick; Levy, Yaron; Massingham, Tim (2009). „Genomová DNA k-mer spektra: modely a modality“. Genome Biology. 10 (10): R108. doi:10.1186 / gb-2009-10-10-r108. ISSN 1465-6906. PMC 2784323. PMID 19814784.
- ^ Yakovchuk, P. (2006-01-30). „Přikládání bází a párování bází k tepelné stabilitě dvojité šroubovice DNA“. Výzkum nukleových kyselin. 34 (2): 564–574. doi:10.1093 / nar / gkj454. ISSN 0305-1048. PMC 1360284. PMID 16449200.
- ^ Bernardi, Giorgio (leden 2000). "Isochores a evoluční genomika obratlovců". Gen. 241 (1): 3–17. doi:10.1016 / S0378-1119 (99) 00485-0. PMID 10607893.
- ^ Hurst, Laurence D .; Obchodník, Alexa R. (03.03.2001). „Vysoký obsah guaninu a cytosinu není adaptací na vysokou teplotu: srovnávací analýza mezi prokaryoty“. Sborník Královské společnosti B: Biologické vědy. 268 (1466): 493–497. doi:10.1098 / rspb.2000.1397. ISSN 1471-2954. PMC 1088632. PMID 11296861.
- ^ A b C Mugal, Carina F .; Weber, Claudia C .; Ellegren, Hans (prosinec 2015). „Konverze genů ovlivněných GC spojuje rekombinační krajinu a demografii se složením genomové báze: Konverze genů ovlivněná GC řídí složení genomové báze u široké škály druhů. BioEssays. 37 (12): 1317–1326. doi:10.1002 / bies.201500058. PMID 26445215. S2CID 21843897.
- ^ Romiguier, Jonathan; Roux, Camille (2017-02-15). „Analytické předsudky spojené s obsahem GC v molekulární evoluci“. Frontiers in Genetics. 8: 16. doi:10.3389 / fgene.2017.00016. ISSN 1664-8021. PMC 5309256. PMID 28261263.
- ^ Spencer, C.C.A. (2006-08-01). "Lidský polymorfismus kolem hotspotů rekombinace: obrázek 1". Transakce s biochemickou společností. 34 (4): 535–536. doi:10.1042 / BST0340535. ISSN 0300-5127. PMID 16856853.
- ^ Weber, Claudia C; Boussau, Bastien; Romiguier, Jonathan; Jarvis, Erich D; Ellegren, Hans (prosinec 2014). „Důkazy o konverzi genu ovlivněného GC jako hybatele rozdílů mezi liniemi ve složení ptačí základny“. Genome Biology. 15 (12): 549. doi:10.1186 / s13059-014-0549-1. ISSN 1474-760X. PMC 4290106. PMID 25496599.
- ^ Lassalle, Florent; Périan, Séverine; Bataillon, Thomas; Nesme, Xavier; Duret, Laurent; Daubin, Vincent (06.02.2015). Petrov, Dmitri A. (ed.). „Evoluce obsahu GC v bakteriálních genech: rozšiřuje se hypotéza zkreslení genů“. Genetika PLOS. 11 (2): e1004941. doi:10.1371 / journal.pgen.1004941. ISSN 1553-7404. PMC 4450053. PMID 25659072.
- ^ Santoyo, G; Romero, D (duben 2005). "Genová konverze a společná evoluce v bakteriálních genomech". Recenze mikrobiologie FEMS. 29 (2): 169–183. doi:10.1016 / j.femsre.2004.10.004. PMID 15808740.
- ^ Bhérer, Claude; Auton, Adam (16. června 2014), John Wiley & Sons Ltd (ed.), „Biased Gene Conversion and its Impact on Genome Evolution“, eLS, John Wiley & Sons, Ltd, doi:10.1002 / 9780470015902.a0020834.pub2, ISBN 9780470015902
- ^ A b Karlin, Samuel (říjen 1998). "Globální dinukleotidové podpisy a analýza genomové heterogenity". Současný názor v mikrobiologii. 1 (5): 598–610. doi:10.1016 / S1369-5274 (98) 80095-7. PMID 10066522.
- ^ Beutler, E .; Gelbart, T .; Han, J. H .; Koziol, J. A .; Beutler, B. (01.01.1989). „Vývoj genomu a genetického kódu: selekce na úrovni dinukleotidů methylací a polyribonukleotidovým štěpením“. Sborník Národní akademie věd. 86 (1): 192–196. Bibcode:1989PNAS ... 86..192B. doi:10.1073 / pnas.86.1.192. ISSN 0027-8424. PMC 286430. PMID 2463621.
- ^ Di Giallonardo, Francesca; Schlub, Timothy E .; Shi, Mang; Holmes, Edward C. (2017-04-15). Dermody, Terence S. (ed.). „Složení dinukleotidů ve zvířecích virech RNA je formováno více rodinou virů než hostitelskými druhy“. Journal of Virology. 91 (8). doi:10.1128 / JVI.02381-16. ISSN 0022-538X. PMC 5375695. PMID 28148785.
- ^ Żemojtel, Tomasz; kiełbasa, Szymon M .; Arndt, Peter F .; Behrens, Sarah; Bourque, Guillaume; Vingron, Martin (01.01.2011). „CpG deaminace vytváří stránky s transkripčními faktory vázajícími s vysokou účinností“. Biologie genomu a evoluce. 3: 1304–1311. doi:10.1093 / gbe / evr107. ISSN 1759-6653. PMC 3228489. PMID 22016335.
- ^ A b Hershberg, R; Petrov, DA (2008). "Výběr na Codon Bias". Výroční přehled genetiky. 42: 287–299. doi:10.1146 / annurev.genet.42.110807.091442. PMID 18983258.
- ^ Sharp, Paul M .; Li, Wen-Hsiung (1987). „Index adaptace kodonů - míra směrového synonymního zkreslení použití kodonů a jeho potenciální aplikace“. Výzkum nukleových kyselin. 15 (3): 1281–1295. doi:10.1093 / nar / 15.3.1281. ISSN 0305-1048. PMC 340524. PMID 3547335.
- ^ Noble, Peter A .; Citek, Robert W .; Ogunseitan, Oladele A. (duben 1998). "Frekvence tetranukleotidů v mikrobiálních genomech". Elektroforéza. 19 (4): 528–535. doi:10,1002 / elps.1150190412. ISSN 0173-0835. PMID 9588798.
- ^ Nagarajan, Niranjan; Pop, Mihai (2013). "Sestava sekvence demystifikována". Genetika hodnocení přírody. 14 (3): 157–167. doi:10.1038 / nrg3367. ISSN 1471-0056. PMID 23358380. S2CID 3519991.
- ^ Li; et al. (2010). „Sestavování lidských genomů de novo s masivně paralelním sekvenováním krátkého čtení“. Výzkum genomu. 20 (2): 265–272. doi:10.1101 / gr.097261.109. PMC 2813482. PMID 20019144.
- ^ Compeau, P .; Pevzner, P .; Teslar, G. (2011). „Jak aplikovat de Bruijnovy grafy na genomové seskupení“. Přírodní biotechnologie. 29 (11): 987–991. doi:10.1038 / nbt.2023. PMC 5531759. PMID 22068540.
- ^ Phillippy, Schatz, Pop (2008). „Forenzní analýza genomu: hledání nepolapitelného chybného sestavení“. Bioinformatika. 9 (3): R55. doi:10.1186 / gb-2008-9-3-r55. PMC 2397507. PMID 18341692.CS1 maint: více jmen: seznam autorů (odkaz)
- ^ Delmont, Eren (2016). „Identifikace kontaminace pomocí pokročilých postupů vizualizace a analýzy: metagenomické přístupy pro sestavy eukaryotických genomů“. PeerJ. 4: e1839. doi:10,7717 / peerj.1839. PMC 4824900. PMID 27069789.
- ^ Bemm; et al. (2016). „Genard tardigradu: Horizontální přenos genů nebo bakteriální kontaminace?“. Sborník Národní akademie věd. 113 (22): E3054 – E3056. doi:10.1073 / pnas.1525116113. PMC 4896698. PMID 27173902.
- ^ A b Zerbino, Daniel R .; Birney, Ewan (2008). "Velvet: algoritmy pro de novo sestavení krátkého čtení pomocí grafů de Bruijn". Výzkum genomu. 18 (5): 821–829. doi:10.1101 / gr.074492.107. PMC 2336801. PMID 18349386.
- ^ Goodur, Haswanee D .; Ramtohul, Vyasanand; Baichoo, Shakuntala (11. 11. 2012). „GIDT - Nástroj pro identifikaci a vizualizaci genomových ostrovů v prokaryotických organismech“. 2012 IEEE 12. mezinárodní konference o bioinformatice a bioinženýrství (BIBE): 58–63. doi:10.1109 / bibe.2012.6399707. ISBN 978-1-4673-4358-9. S2CID 6368495.
- ^ Jaron, K. S .; Moravec, J. C .; Martinkova, N. (2014-04-15). „SigHunt: vyhledávač horizontálního přenosu genů optimalizovaný pro eukaryotické genomy“. Bioinformatika. 30 (8): 1081–1086. doi:10.1093 / bioinformatika / btt727. ISSN 1367-4803. PMID 24371153.
- ^ Starr, M. P .; Mandel, M. (01.04.1969). „DNA Base Composition and Taxonomy of Phytopathogenic and Other Enterobacteria“. Journal of General Microbiology. 56 (1): 113–123. doi:10.1099/00221287-56-1-113. ISSN 0022-1287. PMID 5787000.
- ^ Moore, W. E. C .; Stackebrandt, E .; Kandler, O .; Colwell, R. R .; Krichevsky, M. I .; Truper, H. G .; Murray, R. G. E .; Wayne, L. G .; Grimont, P. A. D. (01.10.1987). „Zpráva ad hoc výboru pro usmíření přístupů k bakteriální systematice“. International Journal of Systematic and Evolutionary Microbiology. 37 (4): 463–464. doi:10.1099/00207713-37-4-463. ISSN 1466-5026.
- ^ Patro, Mount, Kingsford (2014). „Sailfish umožňuje kvantifikaci izoformy bez zarovnání ze čtení RNA-sek pomocí lehkých algoritmů“. Přírodní biotechnologie. 32 (5): 462–464. arXiv:1308.3700. doi:10.1038 / nbt.2862. PMC 4077321. PMID 24752080.CS1 maint: více jmen: seznam autorů (odkaz)
- ^ Navarro-Gomez; et al. (2015). „Phy-Mer: nový srovnávací a na referenci nezávislý klasifikátor mitochondriální haploskupiny“. Bioinformatika. 31 (8): 1310–1312. doi:10.1093 / bioinformatika / btu825. PMC 4393525. PMID 25505086.
- ^ Wang, Rong; Xu, Yong; Liu, Bin (2016). „Identifikace rekombinačního bodu na základě mezerových k-merů“. Vědecké zprávy. 6 (1): 23934. Bibcode:2016NatSR ... 623934W. doi:10.1038 / srep23934. ISSN 2045-2322. PMC 4814916. PMID 27030570.
- ^ Hozza, Michal; Vinař, Tomáš; Brejová, Broňa (2015), Iliopoulos, Costas; Puglisi, Simon; Yilmaz, Emine (eds.), "Jak velký je ten genom? Odhad velikosti a pokrytí genomu z k-mer Abundance Spectra", Zpracování řetězce a načítání informacíSpringer International Publishing, 9309, str. 199–209, doi:10.1007/978-3-319-23826-5_20, ISBN 9783319238258
- ^ Lamichhaney, Sangeet; Fan, Guangyi; Widemo, Fredrik; Gunnarsson, Ulrika; Thalmann, Doreen Schwochow; Hoeppner, Marc P; Kerje, Susanne; Gustafson, Ulla; Shi, Chengcheng (2016). „Strukturální genomové změny jsou základem alternativních reprodukčních strategií v ruffu (Philomachus pugnax)“. Genetika přírody. 48 (1): 84–88. doi:10,1038 / ng.3430. ISSN 1061-4036. PMID 26569123.
- ^ Chae; et al. (2013). „Srovnávací analýza využívající vzory K-mer a K-flank poskytuje důkazy o vývoji sekvence ostrovů CpG v genomech savců“. Výzkum nukleových kyselin. 41 (9): 4783–4791. doi:10.1093 / nar / gkt144. PMC 3643570. PMID 23519616.
- ^ Mohamed Hashim, Abdullah (2015). „Vzácná k-mer DNA: Identifikace sekvenčních motivů a predikce CpG ostrova a promotoru“. Journal of Theoretical Biology. 387: 88–100. doi:10.1016 / j.jtbi.2015.09.014. PMID 26427337.
- ^ Price, Jones, Pevzner (2005). „De novo identifikace opakujících se rodin ve velkých genomech“. Bioinformatika. 21 (supp 1): i351–8. doi:10.1093 / bioinformatika / bti1018. PMID 15961478.CS1 maint: více jmen: seznam autorů (odkaz)
- ^ Meher, Prabina Kumar; Sahu, Tanmaya Kumar; Rao, A.R. (2016). "Identifikace druhů na základě čárového kódu DNA pomocí vektoru funkcí k-mer a klasifikátoru náhodných lesů". Gen. 592 (2): 316–324. doi:10.1016 / j.gene.2016.07.010. PMID 27393648.
- ^ Newburger, Bulyk (2009). „UniPROBE: online databáze dat mikropolí vázajících proteiny o interakcích protein – DNA“. Výzkum nukleových kyselin. 37 (supp 1) (vydání databáze): D77–82. doi:10.1093 / nar / gkn660. PMC 2686578. PMID 18842628.
- ^ Nordstrom; et al. (2013). „Identifikace mutace přímým porovnáním dat sekvenování celého genomu od jedinců mutantního a divokého typu pomocí k-mers“. Přírodní biotechnologie. 31 (4): 325–330. doi:10.1038 / nbt.2515. PMID 23475072.
- ^ Zhu, Jianfeng; Zheng, Wei-Mou (2014). „Samoorganizující se přístup pro meta-genomy“. Výpočetní biologie a chemie. 53: 118–124. doi:10.1016 / j.compbiolchem.2014.08.016. PMID 25213854.
- ^ Dubinkina; Ischenko; Ulyantsev; Tyakht; Alexejev (2016). „Posouzení použitelnosti k-merového spektra pro metagenomickou analýzu odlišnosti“. BMC bioinformatika. 17: 38. doi:10.1186 / s12859-015-0875-7. PMC 4715287. PMID 26774270.
- ^ Teeling, H; Waldmann, J; Lombardot, T; Bauer, M; Glöckner, F (2004). „TETRA: webová služba a samostatný program pro analýzu a srovnání vzorců používání tetranukleotidů v sekvencích DNA“. BMC bioinformatika. 5: 163. doi:10.1186/1471-2105-5-163. PMC 529438. PMID 15507136.
- ^ Chatterji, Sourav; Yamazaki, Ichitaro; Bai, Zhaojun; Eisen, Jonathan A. (2008), Vingron, Martin; Wong, Limsoon (eds.), „CompostBin: Algoritmus založený na složení DNA pro čtení Binningových environmentálních brokovnic“, Výzkum v oblasti výpočetní molekulární biologieSpringer Berlin Heidelberg, 4955, s. 17–28, arXiv:0708.3098, doi:10.1007/978-3-540-78839-3_3, ISBN 9783540788386, S2CID 7832512
- ^ Zheng, Hao; Wu, Hongwei (2010). "Krátké prokaryotické sdružování fragmentů DNA pomocí hierarchického klasifikátoru založeného na lineární diskriminační analýze a analýze hlavních komponent". Journal of Bioinformatics and Computational Biology. 08 (6): 995–1011. doi:10.1142 / S0219720010005051. ISSN 0219-7200. PMID 21121023.
- ^ McHardy, Alice Carolyn; Martín, Héctor García; Tsirigos, Aristotelis; Hugenholtz, Philip; Rigoutsos, Isidore (2007). "Přesná fylogenetická klasifikace fragmentů DNA s proměnnou délkou". Přírodní metody. 4 (1): 63–72. doi:10.1038 / nmeth976. ISSN 1548-7091. PMID 17179938. S2CID 28797816.
- ^ Ounit, Rachid; Wanamaker, Steve; Zavřít, Timothy J; Lonardi, Stefano (2015). „CLARK: rychlá a přesná klasifikace metagenomických a genomických sekvencí pomocí diskriminačních k-merů“. BMC Genomics. 16 (1): 236. doi:10.1186 / s12864-015-1419-2. ISSN 1471-2164. PMC 4428112. PMID 25879410.
- ^ Diaz, Naryttza N; Krause, Lutz; Goesmann, Alexander; Niehaus, Karsten; Nattkemper, Tim W (2009). „TACOA - Taxonomická klasifikace environmentálních genomových fragmentů pomocí přístupu kernelovaného nejbližšího souseda“. BMC bioinformatika. 10 (1): 56. doi:10.1186/1471-2105-10-56. ISSN 1471-2105. PMC 2653487. PMID 19210774.
- ^ Fiannaca, Antonino; La Paglia, Laura; La Rosa, Massimo; Lo Bosco, Giosue “; Renda, Giovanni; Rizzo, Riccardo; Gaglio, Salvatore; Urso, Alfonso (2018). „Modely hloubkového učení pro taxonomickou klasifikaci metagenomických dat bakterií“. BMC bioinformatika. 19 (S7): 198. doi:10.1186 / s12859-018-2182-6. ISSN 1471-2105. PMC 6069770. PMID 30066629.
- ^ Zhu, Zheng (2014). „Samoorganizující se přístup pro meta-genomy“. Výpočetní biologie a chemie. 53: 118–124. doi:10.1016 / j.compbiolchem.2014.08.016. PMID 25213854.
- ^ Lu, Jennifer; Breitwieser, Florian P .; Thielen, Peter; Salzberg, Steven L. (01.01.2017). „Bracken: odhad množství druhů v metagenomických datech“. PeerJ Computer Science. 3: e104. doi:10,7717 / peerj-cs.104. ISSN 2376-5992.
- ^ Wood, Derrick E; Salzberg, Steven L (2014). „Kraken: ultrarychlá klasifikace metagenomických sekvencí s použitím přesných zarovnání“. Genome Biology. 15 (3): R46. doi:10.1186 / gb-2014-15-3-r46. ISSN 1465-6906. PMC 4053813. PMID 24580807.
- ^ Rosen, Gail; Garbarine, Elaine; Caseiro, Diamantino; Polikar, Robi; Sokhansanj, Bahrad (2008). "Klasifikace fragmentů metagenomu pomocí profilů -Mer frekvence". Pokroky v bioinformatice. 2008: 205969. doi:10.1155/2008/205969. ISSN 1687-8027. PMC 2777009. PMID 19956701.
- ^ Wang, Ying; Fu, Lei; Ren, Jie; Yu, Zhaoxia; Chen, Ting; Sun, Fengzhu (03.05.2018). „Identifikace skupinově specifických sekvencí pro mikrobiální komunity pomocí podpisů dlouhých k-mer sekvencí“. Hranice v mikrobiologii. 9: 872. doi:10.3389 / fmicb.2018.00872. ISSN 1664-302X. PMC 5943621. PMID 29774017.
- ^ Al-Saif, Maher; Khabar, Khalid SA (2012). „Snížení frekvence dinukleotidů UU / UA v kódujících oblastech vede ke zvýšené stabilitě mRNA a expresi proteinů“. Molekulární terapie. 20 (5): 954–959. doi:10.1038 / mt.2012.29. PMC 3345983. PMID 22434136.
- ^ Trinh, R; Gurbaxani, B; Morrison, SL; Seyfzadeh, M (2004). "Optimalizace použití kodonových párů v rámci (GGGGS) 3 linkerové sekvence vede ke zvýšené expresi proteinu". Molekulární imunologie. 40 (10): 717–722. doi:10.1016 / j.molimm.2003.08.006. PMID 14644097.
- ^ Shen, Sam H .; Stauft, Charles B .; Gorbatsevych, Oleksandr; Song, Yutong; Ward, Charles B .; Yurovsky, Alisa; Mueller, Steffen; Futcher, Bruce; Wimmer, Eckard (14.04.2015). „Rozsáhlé překódování arbovirového genomu k vyvážení jeho preference hmyzu proti savcům“. Sborník Národní akademie věd. 112 (15): 4749–4754. Bibcode:2015PNAS..112.4749S. doi:10.1073 / pnas.1502864112. ISSN 0027-8424. PMC 4403163. PMID 25825721.
- ^ Kaplan, Bryan S .; Souza, Carine K .; Gauger, Phillip C .; Stauft, Charles B .; Robert Coleman, J .; Mueller, Steffen; Vincent, Amy L. (2018). „Očkování prasat pomocí optimalizované živé oslabené vakcíny proti chřipce zkreslením kodonových párů chrání před homologní výzvou“. Vakcína. 36 (8): 1101–1107. doi:10.1016 / j.vakcina.2018.01.027. PMID 29366707.
- ^ Eschke, Kathrin; Trimpert, Jakob; Osterrieder, Nikolaus; Kunec, Dušan (29.01.2018). Mocarski, Edward (ed.). „Útlum velmi virulentního herpesviru Marekovy choroby (MDV) deoptimizací předpětí kodonových párů“. PLOS patogeny. 14 (1): e1006857. doi:10.1371 / journal.ppat.1006857. ISSN 1553-7374. PMC 5805365. PMID 29377958.
- ^ Kunec, Dušan; Osterrieder, Nikolaus (2016). „Předpětí dvojice kodonů je přímým důsledkem předpětí dinukleotidů“. Zprávy buněk. 14 (1): 55–67. doi:10.1016 / j.celrep.2015.12.011. PMID 26725119.
- ^ Tulloch, Fiona; Atkinson, Nicky J; Evans, David J; Ryan, Martin D; Simmonds, Peter (09.12.2014). „Útlum viru RNA deoptimizací párem kodonů je artefaktem zvýšení frekvencí dinukleotidů CpG / UpA“. eLife. 3: e04531. doi:10,7554 / eLife.04531. ISSN 2050-084X. PMC 4383024. PMID 25490153.
- ^ Marçais, Guillaume; Kingsford, Carl (15.03.2011). „Rychlý přístup bez zámku pro efektivní paralelní počítání výskytů k-merů“. Bioinformatika. 27 (6): 764–770. doi:10.1093 / bioinformatika / btr011. ISSN 1460-2059. PMC 3051319. PMID 21217122.
- ^ Deorowicz, Sebastian; Kokot, Marek; Grabowski, Szymon; Debudaj-Grabysz, Agnieszka (2015-05-15). „KMC 2: rychlé a úsporné počítání k-mer“. Bioinformatika. 31 (10): 1569–1576. doi:10.1093 / bioinformatika / btv022. ISSN 1460-2059. PMID 25609798.
- ^ Erbert, Marius; Rechner, Steffen; Müller-Hannemann, Matthias (2017). „Gerbil: rychlý a paměťově efektivní čítač k-mer s podporou GPU“. Algoritmy pro molekulární biologii. 12 (1): 9. doi:10.1186 / s13015-017-0097-9. ISSN 1748-7188. PMC 5374613. PMID 28373894.