Virtuální projekce - Virtual screening
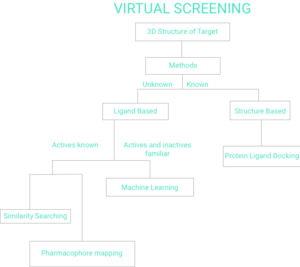
Virtuální projekce (VS) je výpočetní technika používaná v objev drog prohledávat knihovny malé molekuly za účelem identifikace struktur, které se s největší pravděpodobností vážou na a drogový cíl, typicky a protein receptor nebo enzym.[2][3]
Virtuální screening byl definován jako „automatické hodnocení velmi velkých knihoven sloučenin“ pomocí počítačových programů.[4] Jak tato definice naznačuje, VS byla do značné míry hra čísel, která se zaměřuje na to, jak obrovská chemický prostor více než 1060 myslitelné sloučeniny[5] lze filtrovat na zvládnutelné číslo, které lze syntetizovat, zakoupit a otestovat. Ačkoli prohledávání celého chemického vesmíru může být teoreticky zajímavým problémem, praktičtější scénáře VS se zaměřují na návrh a optimalizaci cílených kombinatorických knihoven a obohacení knihoven dostupných sloučenin z interních úložišť sloučenin nebo nabídek prodejců. Jak se zvýšila přesnost metody, virtuální screening se stal nedílnou součástí objev drog proces.[6][1] Virtuální screening lze použít k výběru interních databázových sloučenin pro screening, výběru sloučenin, které lze zakoupit externě, a k výběru, která sloučenina by měla být syntetizována jako další.
Metody
Existují dvě široké kategorie screeningových technik: ligandové a strukturní.[7] Zbytek této stránky bude odrážet vývojový diagram virtuálního screeningu na obrázku 1.
Ligandové
Vzhledem k souboru strukturálně různorodých ligandy který se váže na a receptor, model receptoru lze vytvořit využitím kolektivní informace obsažené v takové sadě ligandů. Tito jsou známí jako farmakofor modely. Kandidátský ligand lze poté porovnat s farmakoforovým modelem, aby se určilo, zda je s ním kompatibilní a proto se pravděpodobně váže.[8]
Dalším přístupem k virtuálnímu screeningu založenému na ligandu je použití 2D metod analýzy chemické podobnosti[9] skenovat databázi molekul proti jedné nebo více aktivním strukturám ligandů.
Populární přístup k virtuálnímu skríningu založenému na ligandu je založen na prohledávání molekul s podobným tvarem jako u známých aktivních látek, protože takové molekuly zapadnou do vazebného místa cíle a budou tedy pravděpodobně vázat cíl. V literatuře existuje řada perspektivních aplikací této třídy technik.[10][11][12] Farmakoforická rozšíření těchto 3D metod jsou také volně dostupná jako webové servery.[13][14]
Na základě struktury
Strukturovaný virtuální screening zahrnuje dokování kandidátských ligandů do proteinového cíle s následnou aplikací a bodovací funkce odhadnout pravděpodobnost, že se ligand bude vázat na protein s vysokou afinitou.[15][16][17] Webové servery zaměřené na budoucí virtuální screening jsou k dispozici všem.[18][19]
Hybridní metody
Byly také vyvinuty hybridní metody, které se spoléhají na strukturní a ligandovou podobnost, aby překonaly omezení tradičních přístupů VLS. Tato metodologie využívá evoluční informace o vazbě ligandů k predikci vazeb malých molekul[20][21] a mohou využívat jak globální strukturální podobnost, tak kapesní podobnost.[20] Přístup založený na globální strukturální podobnosti využívá k nalezení strukturní podobnosti s proteiny v knihovně holo-templátu PDB jak experimentální strukturu, tak model predikovaného proteinu. Po detekci významné strukturální podobnosti se na testování malých molekul, které jsou podobné ligandům extrahovaným z vybraných holo PDB šablon, použije metrika Tanimotova koeficientu založená na 2D otisku prstu.[22][23] Předpovědi z této metody byly experimentálně hodnoceny a ukazují dobré obohacení při identifikaci aktivních malých molekul.
Výše uvedená metoda závisí na globální strukturální podobnosti a není schopna a priori výběr konkrétního místa vázajícího ligand v požadovaném proteinu. Dále, protože se metody spoléhají na posouzení 2D podobnosti pro ligandy, nejsou schopné rozpoznat stereochemickou podobnost malých molekul, které jsou podstatně odlišné, ale prokazují podobnost geometrických tvarů. K řešení těchto obav je nový přístup zaměřený na kapsy, PoLi, schopný cílit na specifické vazebné kapsy v šablonách s holoproteiny, byl vyvinut a experimentálně hodnocen.
Výpočetní infrastruktura
Výpočet párových interakcí mezi atomy, který je předpokladem pro provoz mnoha virtuálních screeningových programů, je výpočetní složitost, kde N je počet atomů v systému. Kvůli kvadratickému škálování s ohledem na počet atomů se výpočetní infrastruktura může lišit od přenosného počítače pro metodu založenou na ligandu po sálového počítače pro metodu založenou na struktuře.

Ligandové
Metody založené na ligandu obvykle vyžadují zlomek sekundy pro operaci porovnání jedné struktury. Jediný procesor stačí k provedení velkého screeningu během několika hodin. Lze však paralelně provést několik srovnání, aby se urychlilo zpracování velké databáze sloučenin.
Na základě struktury
Velikost úkolu vyžaduje a paralelní výpočty infrastruktura, například shluk Linux systémy, běžící dávkový procesor pro zpracování práce, jako např Sun Grid Engine nebo točivý moment PBS.
Je zapotřebí prostředek pro zpracování vstupu z velkých složených knihoven. To vyžaduje formu složené databáze, kterou může dotazovat paralelní klastr, dodávající sloučeniny paralelně s různými výpočetními uzly. Komerční databázové stroje mohou být příliš obtížné a vysokorychlostní indexovací stroj, jako například Berkeley DB, může být lepší volbou. Kromě toho nemusí být efektivní spustit jedno srovnání na úlohu, protože doba rozběhu uzlů clusteru by mohla snadno předstihnout množství užitečné práce. Chcete-li tento problém vyřešit, je nutné zpracovat dávky sloučenin v každé úloze clusteru a agregovat výsledky do nějakého druhu souboru protokolu. Po spuštění celého experimentu lze spustit sekundární proces, který slouží k těžbě souborů protokolu a extrakci kandidátů s vysokým skóre.
Přesnost
Cílem virtuálního screeningu je identifikovat molekuly nové chemické struktury, které se vážou na makromolekulární látky cíl zájmu. Úspěch virtuální obrazovky je tedy definován spíše z hlediska hledání zajímavých nových lešení než z celkového počtu zásahů. Interpretace přesnosti virtuálního screeningu by proto měly být zvažovány opatrně. Nízký míry zásahů Zajímavá lešení jsou jednoznačně výhodnější než vysoké míry úspěšnosti již známých lešení.
Většina testů virtuálních screeningových studií v literatuře je retrospektivních. V těchto studiích se výkon techniky VS měří jeho schopností získat malou sadu dříve známých molekul s afinitou k cílovému cíli (aktivní molekuly nebo jen aktivní látky) z knihovny obsahující mnohem vyšší podíl předpokládaných nečinností nebo návnady. Naproti tomu v perspektivních aplikacích virtuálního screeningu jsou výsledné zásahy podrobeny experimentálnímu potvrzení (např. IC50 Měření). Panuje shoda v tom, že retrospektivní referenční hodnoty nejsou dobrým prediktorem budoucího výkonu a v důsledku toho představují pouze prospektivní studie nezvratný důkaz vhodnosti techniky pro konkrétní cíl.[24][25][26][27][28]
Aplikace na objevování drog
Virtuální screening je velmi užitečná aplikace, pokud jde o identifikaci zasažených molekul jako počátek medicínské chemie. Vzhledem k tomu, že přístup virtuálního screeningu se začíná stávat životně důležitější a podstatnější technikou v průmyslu medicinální chemie, tento přístup se rychle zvýšil.[29]
Metody založené na ligandu
I když nezná strukturu, snaží se předpovědět, jak se ligandy budou vázat na receptor. S využitím vlastností farmakoforů každý ligand identifikoval dárce a akceptory. Rovnocenné funkce jsou překryty, ale vzhledem k tomu, že je nepravděpodobné, že existuje jediné správné řešení.[1]
Farmakoforové modely
Tato technika se používá při slučování výsledků vyhledávání pomocí na rozdíl od referenčních sloučenin, stejných deskriptorů a koeficientu, ale různých aktivních sloučenin. Tato technika je výhodná, protože je efektivnější než použití jediné referenční struktury spolu s nejpřesnějším výkonem, pokud jde o různé aktivní složky.[1]
Pharmacophore je soubor sterických a elektronických funkcí, které jsou potřebné k optimální supramolekulární interakci nebo interakcím s biologickou cílovou strukturou, aby se urychlila její biologická odpověď. Vyberte zástupce jako sadu aktivních látek, většina metod bude hledat podobné vazby. Je výhodné mít více tuhých molekul a ligandy by měly být diverzifikovány, jinými slovy zajistit, aby měly různé vlastnosti, které se během vazebné fáze nevyskytují.[1]
Struktura
Vytvořte složený prediktivní model založený na známých aktivních a známých neaktivních znalostech. QSAR (vztah kvantitativní struktury a aktivity), který je omezen na malou homogenní datovou sadu. SAR (Structure Activity Relationship), kde se s daty zachází kvalitativně a lze je použít se strukturálními třídami a více než jedním režimem vazby. Modely upřednostňují sloučeniny pro objev olova.[1]
Strojové učení
Aby bylo možné použít Machine Learning pro tento model virtuálního screeningu, musí existovat tréninková sada se známými aktivními a známými neaktivními sloučeninami. K dispozici je také model aktivity, který je poté vypočítán pomocí substrukturální analýzy, rekurzivního dělení, podpory vektorových strojů, k nejbližším sousedům a neuronových sítí. Posledním krokem je zjištění pravděpodobnosti, že je sloučenina aktivní, a následné seřazení každé sloučeniny na základě její pravděpodobnosti, že bude aktivní.[1]
Substrukturní analýza ve strojovém učení
Prvním modelem strojového učení používaným na velkých datových sadách je analýza substruktury, která byla vytvořena v roce 1973. Každá substruktura fragmentu neustále přispívá k aktivitě konkrétního typu.[1] Substruktura je metoda, která překonává obtíže masivní dimenzionality, pokud jde o analýzu struktur v designu léků. Efektivní analýza spodní stavby se používá pro konstrukce, které mají podobnost s víceúrovňovou budovou nebo věží. Geometrie se používá k číslování mezních spojů pro danou konstrukci na začátku a směrem k vyvrcholení. Při vývoji metody speciální statické kondenzace a substitučních rutin se ukázalo, že tato metoda je produktivnější než předchozí modely analýzy spodní konstrukce.[30]
Rekurzivní rozdělení na oddíly
Rekurzivně dělení je metoda, která vytváří rozhodovací strom pomocí kvalitativních dat. Pochopení způsobu, jakým pravidla rozdělují třídy s malou chybou nesprávné klasifikace, zatímco opakují každý krok, dokud nenajdete žádné rozumné rozdělení. Rekurzivní rozdělení na oddíly však může mít špatnou schopnost predikce a potenciálně vytvářet jemné modely stejnou rychlostí.[1]
Strukturní metody známé dokování proteinových ligandů
Ligand se může vázat na aktivní místo v proteinu pomocí dokovacího vyhledávacího algoritmu a skórovací funkce za účelem identifikace nejpravděpodobnější příčiny individuálního ligandu při přiřazování prioritního pořadí.[1][31]
Viz také
Reference
- ^ A b C d E F G h i j Gillet V (2013). „Virtuální screening založený na ligandech a strukturách“ (PDF). University of Sheffield.
- ^ Rester U (červenec 2008). „Od virtuality k realitě - Virtuální screening při objevování a optimalizaci elektrod: perspektiva léčivé chemie“. Aktuální názor na objev a vývoj drog. 11 (4): 559–68. PMID 18600572.
- ^ Rollinger JM, Stuppner H, Langer T (2008). „Virtuální screening na objev bioaktivních přírodních produktů“. Přírodní sloučeniny jako léky, svazek I. Pokrok ve výzkumu drog. Fortschritte der Arzneimittelforschung. Progres des Recherches Pharmaceutiques. Pokrok ve výzkumu drog. 65. 211, 213–49. doi:10.1007/978-3-7643-8117-2_6. ISBN 978-3-7643-8098-4. PMC 7124045. PMID 18084917.
- ^ Walters WP, Stahl MT, Murcko MA (1998). Msgstr "Virtuální screening - přehled". Drug Discov. Dnes. 3 (4): 160–178. doi:10.1016 / S1359-6446 (97) 01163-X.
- ^ Bohacek RS, McMartin C, Guida WC (1996). „Umění a praxe konstrukce léků na základě struktury: perspektiva molekulárního modelování“. Med. Res. Rev. 16 (1): 3–50. doi:10.1002 / (SICI) 1098-1128 (199601) 16: 1 <3 :: AID-MED1> 3.0.CO; 2-6. PMID 8788213.
- ^ McGregor MJ, Luo Z, Jiang X (11. června 2007). „Kapitola 3: Virtuální screening při objevování drog“. V Huang Z (ed.). Výzkum objevů drog. Nové hranice v postgenomické éře. Wiley-VCH: Weinheim, Německo. str. 63–88. ISBN 978-0-471-67200-5.
- ^ McInnes C (říjen 2007). "Virtuální screeningové strategie při objevování drog". Aktuální názor na chemickou biologii. 11 (5): 494–502. doi:10.1016 / j.cbpa.2007.08.033. PMID 17936059.
- ^ Sun H (2008). „Virtuální screening založený na farmakoforech“. Současná léčivá chemie. 15 (10): 1018–24. doi:10.2174/092986708784049630. PMID 18393859.
- ^ Willet P, Barnard JM, Downs GM (1998). "Hledání chemické podobnosti". Journal of Chemical Information and Computer Sciences. 38 (6): 983–996. CiteSeerX 10.1.1.453.1788. doi:10.1021 / ci9800211.
- ^ Rush TS, Grant JA, Mosyak L, Nicholls A (březen 2005). „Tvarově založená metoda skákání 3-D lešení a její aplikace na interakci bakteriální protein-protein“. Journal of Medicinal Chemistry. 48 (5): 1489–95. CiteSeerX 10.1.1.455.4728. doi:10.1021 / jm040163o. PMID 15743191.
- ^ Ballester PJ, Westwood I, Laurieri N, Sim E, Richards WG (únor 2010). „Perspektivní virtuální screening s ultrarychlým rozpoznáváním tvaru: identifikace nových inhibitorů arylaminových N-acetyltransferáz“. Journal of the Royal Society, Interface. 7 (43): 335–42. doi:10.1098 / rsif.2009.0170. PMC 2842611. PMID 19586957.
- ^ Kumar A, Zhang KY (2018). „Pokroky ve vývoji metod tvarové podobnosti a jejich aplikace při objevování drog“. Hranice v chemii. 6: 315. Bibcode:2018FrCh .... 6..315K. doi:10.3389 / fchem.2018.00315. PMC 6068280. PMID 30090808.
- ^ Li H, Leung KS, Wong MH, Ballester PJ (červenec 2016). „USR-VS: webový server pro perspektivní virtuální screening ve velkém měřítku pomocí ultrarychlých technik rozpoznávání tvarů“. Výzkum nukleových kyselin. 44 (W1): W436–41. doi:10.1093 / nar / gkw320. PMC 4987897. PMID 27106057.
- ^ Sperandio O, Petitjean M, Tuffery P (červenec 2009). „wwLigCSRre: server založený na 3D ligandu pro identifikaci a optimalizaci zásahu“. Výzkum nukleových kyselin. 37 (Problém s webovým serverem): W504–9. doi:10.1093 / nar / gkp324. PMC 2703967. PMID 19429687.
- ^ Kroemer RT (srpen 2007). „Strukturovaný design léků: dokování a hodnocení“. Současná věda o proteinech a peptidech. 8 (4): 312–28. CiteSeerX 10.1.1.225.959. doi:10.2174/138920307781369382. PMID 17696866.
- ^ Cavasotto CN, Orry AJ (2007). "Ligandové dokování a strukturní virtuální screening v objevování drog". Aktuální témata v medicinální chemii. 7 (10): 1006–14. doi:10.2174/156802607780906753. PMID 17508934.
- ^ Kooistra AJ, Vischer HF, McNaught-Flores D, Leurs R, de Esch IJ, de Graaf C (2016). „Funkčně specifický virtuální screening na GPCR ligandy pomocí kombinované metody skórování“. Vědecké zprávy. 6: 28288. Bibcode:2016NatSR ... 628288K. doi:10.1038 / srep28288. PMC 4919634. PMID 27339552.
- ^ Irwin JJ, Shoichet BK, Mysinger MM, Huang N, Colizzi F, Wassam P, Cao Y (září 2009). „Automatizované dokovací obrazovky: studie proveditelnosti“. Journal of Medicinal Chemistry. 52 (18): 5712–20. doi:10.1021 / jm9006966. PMC 2745826. PMID 19719084.
- ^ Li H, Leung KS, Ballester PJ, Wong MH (2014-01-24). „istar: webová platforma pro dokování rozsáhlých proteinových ligandů“. PLOS ONE. 9 (1): e85678. Bibcode:2014PLoSO ... 985678L. doi:10.1371 / journal.pone.0085678. PMC 3901662. PMID 24475049.
- ^ A b Zhou H, Skolnick J (leden 2013). „FINDSITE (comb): threading / structure-based, proteomic-scale virtual approach approach“. Journal of Chemical Information and Modeling. 53 (1): 230–40. doi:10.1021 / ci300510n. PMC 3557555. PMID 23240691.
- ^ Roy A, Skolnick J (únor 2015). „LIGSIFT: open-source nástroj pro strukturální zarovnání ligandu a virtuální screening“. Bioinformatika. 31 (4): 539–44. doi:10.1093 / bioinformatika / btu692. PMC 4325547. PMID 25336501.
- ^ Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP (leden 2012). „ChEMBL: rozsáhlá databáze bioaktivity pro objevování drog“. Výzkum nukleových kyselin. 40 (Problém s databází): D1100–7. doi:10.1093 / nar / gkr777. PMC 3245175. PMID 21948594.
- ^ Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J (leden 2006). „DrugBank: komplexní zdroj pro objevování a průzkum drog in silico“. Výzkum nukleových kyselin. 34 (Problém s databází): D668–72. doi:10.1093 / nar / gkj067. PMC 1347430. PMID 16381955.
- ^ Wallach I, Heifets A (2018). "Většina klasifikačních standardů založených na ligandech odměňuje memorování namísto zobecnění". Journal of Chemical Information and Modeling. 58 (5): 916–932. arXiv:1706.06619. doi:10.1021 / acs.jcim.7b00403. PMID 29698607.
- ^ Irwin JJ (2008). „Referenční hodnoty Společenství pro virtuální screening“. Journal of Computer-Aided Molecular Design. 22 (3–4): 193–9. Bibcode:2008JCAMD..22..193I. doi:10.1007 / s10822-008-9189-4. PMID 18273555. S2CID 26260725.
- ^ Dobrý AC, Oprea TI (2008). „Optimalizace technik CAMD 3. Virtuální screeningové obohacovací studie: pomoc nebo překážka při výběru nástroje?“. Journal of Computer-Aided Molecular Design. 22 (3–4): 169–78. Bibcode:2008JCAMD..22..169G. doi:10.1007 / s10822-007-9167-2. PMID 18188508. S2CID 7738182.
- ^ Schneider G (duben 2010). „Virtuální projekce: nekonečné schodiště?“. Recenze přírody. Objev drog. 9 (4): 273–6. doi:10.1038 / nrd3139. PMID 20357802. S2CID 205477076.
- ^ Ballester PJ (leden 2011). "Ultrarychlé rozpoznávání tvarů: metoda a aplikace". Budoucí léčivá chemie. 3 (1): 65–78. doi:10,4155 / fmc. 10,280. PMID 21428826.
- ^ Lavecchia A, Di Giovanni C (2013). „Virtuální screeningové strategie při objevování drog: kritická recenze“. Současná léčivá chemie. 20 (23): 2839–60. doi:10.2174/09298673113209990001. PMID 23651302.
- ^ Gurujee CS, Deshpande VL (únor 1978). Msgstr "Vylepšená metoda analýzy spodní stavby". Počítače a struktury. 8 (1): 147–152. doi:10.1016/0045-7949(78)90171-2.
- ^ Pradeepkiran, Jangampalli Adi; Reddy, P. Hemachandra (březen 2019). „Strukturovaný design a studie molekulárního dokování pro fosforylované inhibitory Tau u Alzheimerovy choroby“. Buňky. 8 (3): 260. doi:10,3390 / buňky8030260. PMC 6468864. PMID 30893872.
Další čtení
- Melagraki G, Afantitis A, Sarimveis H, Koutentis PA, Markopoulos J, Igglessi-Markopoulou O (květen 2007). „Optimalizace antagonistů MCH1 receptoru biarylpiperidinu a 4-amino-2-biarylmočoviny pomocí QSAR modelování, klasifikačních technik a virtuálního screeningu“. Journal of Computer-Aided Molecular Design. 21 (5): 251–67. Bibcode:2007JCAMD..21..251M. doi:10.1007 / s10822-007-9112-4. PMID 17377847. S2CID 19563229.
- Afantitis A, Melagraki G, Sarimveis H, Koutentis PA, Markopoulos J, Igglessi-Markopoulou O (únor 2006). "Zkoumání substitučního účinku 1- (3,3-difenylpropyl) -piperidinyl-fenylacetamidu na vazebnou afinitu k CCR5 pomocí QSAR a technik virtuálního screeningu". Journal of Computer-Aided Molecular Design. 20 (2): 83–95. Bibcode:2006JCAMD..20 ... 83A. CiteSeerX 10.1.1.716.8148. doi:10.1007 / s10822-006-9038-2. PMID 16783600. S2CID 21523436.
- Eckert H, Bajorath J (březen 2007). "Analýza molekulární podobnosti ve virtuálním screeningu: základy, omezení a nové přístupy". Objev drog dnes. 12 (5–6): 225–33. doi:10.1016 / j.drudis.2007.01.011. PMID 17331887.
- Willett P (prosinec 2006). „Virtuální screening založený na podobnosti pomocí 2D otisků prstů“ (PDF). Objev drog dnes (Vložený rukopis). 11 (23–24): 1046–53. doi:10.1016 / j.drudis.2006.10.005. PMID 17129822.
- Fara DC, Oprea TI, Prossnitz ER, Bologa CG, Edwards BS, Sklar LA (2006). „Integrace virtuálního a fyzického screeningu“. Drug Discovery Today: Technologies. 3 (4): 377–385. doi:10.1016 / j.ddtec.2006.11.003. PMC 7105924.
- Muegge I, Oloffa S (2006). „Pokroky ve virtuálním prověřování“. Drug Discovery Today: Technologies. 3 (4): 405–411. doi:10.1016 / j.ddtec.2006.12.002. PMC 7105922.
- Schneider G (duben 2010). „Virtuální projekce: nekonečné schodiště?“. Recenze přírody. Objev drog. 9 (4): 273–6. doi:10.1038 / nrd3139. PMID 20357802. S2CID 205477076.
externí odkazy
- VLS3D - seznam více než 2000 databází, online i samostatných in silico nástroje