Payne přesmyk - Payne rearrangement
The Payne přesmyk je izomerizace za základních podmínek 2,3-epoxyalkoholů na izomerní 2,3-epoxyalkoholy s obrácenou konfigurací. Aza- a thia-Payneovy přesmyky aziridinů, respektive thiirania, jsou také známy.[1]
Úvod
Za zásadních, protických podmínek procházejí 2,3-epoxyalkoholy přesmykem, při kterém alkoholový kyslík otevírá epoxid s obrácenou konfigurací a vytváří izomerní 1,2-epoxy alkohol. Celkově představuje Payneův přesmyk migraci epoxidu. I když je migrace sama o sobě plně reverzibilní, nukleofilní otevření za Curtin-Hammettových podmínek poskytuje dobré výtěžky funkcionalizovaných diolů odvozených od jediného epoxyalkoholového izomeru.[2] K dokončení reakce lze také použít intramolekulární elektrofilní zachycení nového alkoxidu generovaného při přesmyku. V některých případech je termodynamický rozdíl mezi epoxidovými izomery dostatečně velký na to, aby se získal jediný izomer v synteticky použitelném výtěžku, aniž by se spoléhalo na kinetické rozdíly spojené se zachycením.
(1)

K vyvolání ekvilibrace jsou vyžadovány zásadně základní podmínky, které omezují syntetickou užitečnost transformace na substráty postrádající funkčnost labilní vůči bázím. Mnoho rovnováh epoxidových alkoholů je velmi jemně vyvážených;[3] využití výhod výše popsaných strategií zachycování však může vést k vysokým výtěžkům jednotlivých izomerů.
Mechanismus a stereochemie
Převládající mechanismus
Základní mechanismus Paynova přesmyku zahrnuje deprotonaci volné hydroxylové skupiny, invertivní nukleofilní útok na proximální epoxidový uhlík a opětovnou protonaci nově uvolněného alkoxidu. Každý krok procesu je reverzibilní.[4]
(2)

Několik pozorování naznačuje, že tento mechanistický obraz je příliš zjednodušený. Migrace epoxidu buď nenastává, nebo je za dubotických podmínek velmi pomalá[3]- bylo navrženo, že nukleofilní útok je zpomalen koordinací kovových iontů na nukleofilní kyslík za aprotických podmínek. Navíc, když se k ekvilibračním epoxidovým izomerům přidá externí nukleofil, poměr otevřených produktů neodráží poměr epoxidových izomerů v roztoku nebo jejich relativní termodynamickou stabilitu.[5] In situ příkladem je nukleofilní otevření ekvilibračních epoxidů Curtin-Hammettovy podmínky —Protože epoxidy se rychle vyrovnávají relativně k rychlosti otevírání epoxidu, je to kinetické bariéry otevření kruhu které kontrolují pozorovaný poměr produktu. V níže uvedeném příkladu je produkt otevření terminálního epoxidu hlavním produktem, přestože samotný terminální epoxid je méně termodynamicky stabilní než vnitřní izomer.
(3)

Halo dioly mohou být použity jako prekurzory 2,3-epoxy alkoholů před přesmykem. Problémy se selektivitou místa mohou nastat, pokud dvě hydroxylové skupiny obklopující halogenid nejsou ekvivalentní. Obecně je tvorba vnitřních substituovaných epoxidů rychlejší než tvorba koncových epoxidů.[6] Tuto myšlenku lze použít k předpovědi průběhu migrace in situ-generované epoxidy.
(4)

Stereochemie
Paynův přesmyk nastává s inverzí stereochemie na C-2. Substráty obsahující více sousedních hydroxylových skupin mohou podstoupit „kaskádovou“ migraci epoxidu s inverzí v každém místě nukleofilního napadení. V jednom příkladu dochází po dvou migracích epoxidu, otevření epoxidu karboxylátem a hydrolýze výsledného laktonu k inverzi tří sousedících stereocenter.[7]
(5)

Rozsah a omezení
Payne přesmyk
Pozici rovnováhy v cyklických i acyklických systémech lze předvídat ze struktur dvou rovnovážných epoxidů. V acyklických systémech byla zavedena tato pravidla:[8]
- Upřednostňuje se větší substituce na epoxidovém kruhu.
- Mezi disubstituovanými epoxidy trans izomery jsou upřednostňovány cis izomery.
- Výhodné jsou izomery s primárními hydroxylovými skupinami.
- Substituenty darující elektrony na epoxidu se stabilizují a substituenty přitahující elektrony se destabilizují.
Pyranosidy jsou nejvíce studované cyklické systémy. Studie migrace epoxidů v pyranosidech a jiných cyklických epoxidových alkoholech odhalily tři zevšeobecnění:
- Stejně jako v acyklických systémech je upřednostňována větší substituce na epoxidovém kruhu.
- Preferovaný izomer je ten, který má více pseudoekvatoriálních substituentů.
- Intramolekulární vodíková vazba a další meziprostorové interakce nehrají roli v rovnovážných poměrech.
Konformačně uzamčené pyranosidy odhalují termodynamickou preferenci cyklických substrátů pro více pseudoekvatoriálních skupin.[9]
(6)

Za aprotických podmínek lze nukleofilní otevření epoxidových izomerů dosáhnout hydridy nebo organokupráty. Nukleofilní útok obvykle probíhá na nejméně substituovaném uhlíku, čímž se získá více substituovaný diolový produkt.[10]
(7)

Za protických podmínek je také obvykle upřednostňováno otevření v nejméně substituované poloze. Nukleofily, které mohou být použity za protických podmínek, zahrnují fenoly, sekundární aminy, azidový anion a sulfidy.[11]
(8)

Intermolekulární nukleofilní zachycení jediného epoxidového izomeru je obtížné, protože reakce epoxy alkoholu s elektrofilem je obvykle rychlejší než migrace. Nicméně, uvnitřmolekulární elektrofie jsou často účinné pro zachycení jediného epoxidového izomeru. Například druhý blízký epoxid ve výchozím materiálu rovnice (9) je zachycen jediným epoxidovým izomerem, což vede k tetrahydrofuran.[12]
(9)

Aza- a thia-payne přesmyky
Aza-Paynův přesmyk může být proveden buď ve směru „dopředu“ (epoxid na aziridin) nebo „vzad“ (aziridin na epoxid) v závislosti na použitých podmínkách. Aziridiny chudé na elektrony procházejí reverzním přeskupením v přítomnosti hydridové báze,[13] zatímco odpovídající epoxyaminy procházejí dopředným přesmykem v přítomnosti etherátu fluoridu boritého.[14]
(10)

Přeskupení thia-Payne bylo pozorováno pouze v dopředném směru (epoxid na thiiranium) s in situ otevření thiirania. Inverzní nukleofilní otevření na C-2 je možné za použití trialkylaluminiových reagencií.[15]
(11)

Syntetické aplikace
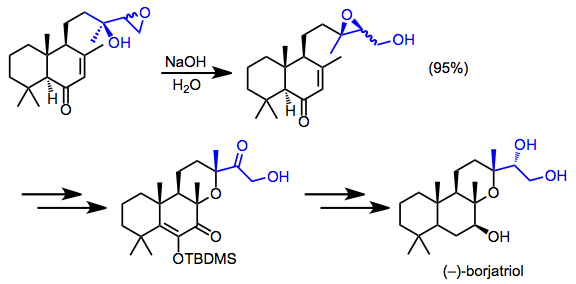
Syntéza borjatriolu zahrnovala vzácnou izolaci migrovaného epoxidu. Po zbytek syntézy byla provedena diastereomerní směs produktů přesmyku.[16]
(12)
Poslední dva kroky v celkové syntéze spatolu zahrnovaly intramolekulární elektrofilní zachycení alkoxidu odvozeného od přeskupeného epoxidu. Útok intermediárního alkoxidu na sousední mesylát poskytl bis (epoxid) a debenzylace poskytla cílovou sloučeninu.[17]
(13)

Srovnání s jinými metodami
Jiné způsoby dostupné pro přípravu 2,3-epoxyalkoholů mají tu výhodu, že nezačínají s existujícím 2,3-epoxy alkoholem; mají však tendenci zahrnovat více kroků než migrace epoxidů. Asymetrická dihydroxylace může být použita k syntéze epoxyalkoholů s vysokou stereoselektivitou a některé metody založené na dihydroxylaci se vyhýbají použití silně bazických podmínek.[18]
(14)

Alternativní metoda, která vede k udržení konfigurace na C-2, zahrnuje mesylaci epoxyalkoholu, otevření epoxidu a opětné uzavření vytěsněním mesylátu.[11]
(15)

Experimentální podmínky a postup
Typické podmínky
Za podmínek přesmyku může dojít k otevření koncových epoxidů náhodným hydroxidem; pokud to není žádoucí, musí se použít bezvodá rozpouštědla, činidla a sklo. Čerstvě připravený methoxid sodný v methanolu se běžně používá k provedení přesmyku bez otevření. Nukleofilního otevírání lze dosáhnout použitím azid sodný, přebytek hydroxidu nebo měďnatých reagentů v přítomnosti chlorid lithný. Elektrofilní zachycování se provádí za standardních podmínek v přítomnosti elektrofilu, jako je benzylbromid. Silylhalogenidy byly také použity jako elektrofilní zachycovací činidla.
Aby se zabránilo migraci epoxidu, lze použít slabě bazické podmínky. Vodný uhličitan draselný ani vodné aminové báze nezpůsobují přeskupení epoxidu. Nízké teploty jsou také výhodné, když není žádoucí migrace epoxidů.
Příklad postupu[19]
(16)

Roztok methyl (kyano) kuprátu (roztok A) se připraví následujícím způsobem: k suspenzi 0,35 g (3,91 mmol) kyanidu měďného v 5 ml tetrahydrofuranu se pod atmosférou argonu při teplotě 0 ° přikape během asi 5 minut 2,76 ml roztoku methyllithia v ethyletheru (1,4 M, 3,86 mmol). Bezbarvý roztok se míchá 10 minut při 0 ° C, během 30 minut se zahřeje na 25 ° C a poté znovu ochladí na 0 ° C. Samostatně se připraví roztok lithné soli (±) -cis-4-benzyloxy-2,3-epoxy-l-butanolu (roztok B) následujícím způsobem: k roztoku 0,5 g (2,58 mmol) epoxidu alkohol a 0,90 g (21,4 mmol) chloridu lithného v 10 ml tetrahydrofuranu pod argonem při -78 ° C bylo po kapkách přidáno 1,65 ml roztoku n-butyllithia v hexanu (1,56 M, 2,58 mmol). Roztok byl míchán po dobu 5 minut při -78 ° C, ponechán ohřát na 0 ° a poté míchán při této teplotě po dobu 10 minut. Reakce byla provedena přidáním roztoku A do roztoku B kanylou při 0 ° C a následným ohřátím na teplotu místnosti po dobu 2 hodin. Reakční směs se poté míchá dalších 12 hodin a poté se opatrně zpracuje s 5 ml nasyceného vodného roztoku chlorid amonný. Směs byla míchána 1–2 hodiny, aby se napomohlo odstranění zbytků mědi. Poté byl přidán ethylether (20 ml) a organická vrstva byla oddělena. Vodná fáze se dvakrát extrahuje 20 ml ethyletheru a spojené organické fáze se vysuší Síran hořečnatý, filtruje se a koncentruje se, čímž se získá 0,51 g produktu jako bezbarvý olej (95%), IR (film) 3400, 3100, 3060, 3030, 2970, 2930, 2870, 1600, 1500, 1465, 1445, 1385, 1370 , 1320, 1285, 1210, 1180, 1120, 1100, 1075, 1030, 1020, 980, 905, 830, 750, 730, 710, 695 cm – 1; 'H NMR (CDCI3) 5 0,90 (t, J = 6,0 Hz, 3 H), 1,37–1,53 (m, 2 H), 3,20 (br s, 2 H), 3,40–3,65 (m, 4 H), 4,48 (s, 2 H) ), 7,29 (s, 5 H).
Reference
- ^ Hanson, R. Org. Reagovat. 2002, 60, 1. doi:10.1002 / 0471264180.nebo060.01
- ^ Seeman, J. I. Chem. Rev. 1983, 83, 83.
- ^ A b Payne, G. B. J. Org. Chem. 1962, 27, 3819.
- ^ Angyal, S. J .; Gilham, P. T. J. Chem. Soc. 1957, 3691.
- ^ Katsuki, T .; Lee, A. W. M .; Ma, P .; Martin, V. S .; Masamune, S .; Sharpless, K. B .; Tuddenham, D .; Walker, F. J. J. Org. Chem. 1982, 47, 1373.
- ^ Paulsen, H .; Eberstein, K. Chem. Ber. 1976, 109, 3891.
- ^ Bock, K .; Lundt, I .; Pedersen, C. Sacharidy. Res. 1988, 179, 87.
- ^ Pierre, J.-L .; Chautemps, P .; Arnaud, P. Býk. Soc. Chim. Fr. 1969, 106, 1317.
- ^ Mubarak, A .; Fraser-Reid, B. J. Org. Chem. 1982, 47, 4265.
- ^ Page, P. C. B .; Rayner, C. M .; Sutherland, I. O. J. Chem. Soc., Perkin Trans. 1 1990, 1375.
- ^ A b Behrens, C.H .; Ko, S. Y .; Sharpless, K. B .; Walker, F. J. J. Org. Chem. 1985, 50, 5687.
- ^ Klein, E .; Rojahn, W .; Henneberg, D. Čtyřstěn 1964, 20, 2025.
- ^ Harden, R. C .; Hodgkinson, T. J .; McKillop, A .; Prowse, W. G .; Urquhart, M. W. J. Čtyřstěn 1997, 53, 21.
- ^ Nakai, K .; Ibuka, T .; Otaka, A .; Tamamura, H .; Fujii, N .; Yamamoto, Y. Tetrahedron Lett. 1995, 36, 6247.
- ^ Sasaki, M .; Tanino, K .; Miyashita, M. J. Org. Chem. 2001, 66, 5388.
- ^ Herlem, D .; Khuonghuu, F. Čtyřstěn 1997, 53, 673.
- ^ Soloman, R.G .; Basu, B .; Roy, S .; Sachinuala, N. D. J. Am. Chem. Soc. 1991, 113, 3096.
- ^ Ko, S. Y .; Malik, M. Tetrahedron Lett. 1993, 34, 4675.
- ^ Page, P. C. B .; Rayner, C. M .; Sutherland, I. O. J. Chem. Soc., Perkin Trans. 1 1990, 1375.