Gibbsova izoterma - Gibbs isotherm
The Gibbsova adsorpční izoterma pro vícesložkové systémy je rovnice používaná k uvádění do vztahu změn koncentrace složky v kontaktu s povrchem se změnami v povrchové napětí, což má za následek odpovídající změnu v povrchová energie. Pro binární systém je Gibbsova adsorpční rovnice z hlediska povrchového přebytku:

kde
- je povrchové napětí,
- i je povrchový přebytek složky i,
- i je chemický potenciál složky i.



Adsorpce
Různé vlivy na rozhraní mohou způsobit změny ve složení blízké povrchové vrstvy.[1] Látky se mohou hromadit blízko povrchu nebo se naopak mohou hromadit.[1] Pohyb molekul charakterizuje jevy adsorpce. Adsorpce ovlivňuje změny v povrchové napětí a koloidní stabilita. Adsorpční vrstvy na povrchu kapalného disperzního média mohou ovlivňovat interakce dispergovaných částic v médiu a následně mohou tyto vrstvy hrát zásadní roli ve stabilitě koloidů[2] K adsorpci molekul kapalné fáze na rozhraní dochází, když je tato kapalná fáze v kontaktu s jinými nemísitelnými fázemi, které mohou být plynné, kapalné nebo pevné[3]
Koncepční vysvětlení rovnice
Povrchové napětí popisuje, jak obtížné je rozšířit plochu povrchu (roztažením nebo deformací). Pokud je povrchové napětí vysoké, je zapotřebí velká volná energie ke zvětšení povrchové plochy, takže povrch bude mít tendenci se smršťovat a držet pohromadě jako pryžový list.
Existuje několik faktorů ovlivňujících povrchové napětí, jedním z nich je, že složení povrchu se může lišit od objemu. Například pokud je voda smíchána s malým množstvím povrchově aktivní látky (například, mýdlo na ruce ), objemová voda může být 99% molekul vody a 1% molekul mýdla, ale nejvyšší povrch vody může být 50% molekul vody a 50% molekul mýdla. V tomto případě má mýdlo velký a pozitivní „povrchový přebytek“. V dalších příkladech může být povrchový přebytek negativní: Například pokud je voda smíchána s anorganická sůl jako chlorid sodný, je povrch vody v průměru méně slané a čistší než hromadný průměr.
Zvažte znovu příklad vody s trochou mýdla. Jelikož vodní povrch musí mít vyšší koncentraci mýdla než objem, vždy, když se zvětší povrch vody, je nutné odstranit molekuly mýdla z objemu a přidat je na nový povrch. Pokud se koncentrace mýdla trochu zvýší, molekuly mýdla jsou snadněji dostupné (mají vyšší chemický potenciál ), takže je snazší je vytáhnout z objemu, aby se vytvořil nový povrch. Jelikož je snazší vytvořit nový povrch, povrchové napětí se sníží. Obecná zásada je:
- Když je povrchový přebytek komponenty pozitivní, zvýšení chemického potenciálu této složky snižuje povrchové napětí.
Dále zvažte příklad vody se solí. Vodní plocha je méně slaná než objemová, takže kdykoli se zvětší povrchová plocha vody, je nutné odstranit molekuly soli z nového povrchu a vtlačit je do objemu. Pokud se koncentrace soli trochu zvýší (zvýšení soli chemický potenciál ), je těžší vytlačit molekuly soli. Protože je nyní těžší vytvořit nový povrch, je povrchové napětí vyšší. Obecná zásada je:
- Když je povrchový přebytek komponenty negativní, zvýšení chemického potenciálu této složky zvyšuje povrchové napětí.
Gibbsova izotermická rovnice poskytuje přesný kvantitativní vztah pro tyto trendy.
Umístění povrchu a definování přebytku povrchu

Umístění povrchu
Za přítomnosti dvou fází (α a β), povrch (povrchová fáze) se nachází mezi fáze α a fáze β. Experimentálně je obtížné určit přesnou strukturu nehomogenní povrchové fáze, která je ve styku s objemovou kapalnou fází obsahující více než jednu rozpuštěnou látku.[2] Nehomogenita povrchové fáze je výsledkem variací molárních poměrů.[1] Model navržený Josiah Willard Gibbs navrhl, aby povrchová fáze jako idealizovaný model, který měl nulovou tloušťku. Ve skutečnosti, i když většina regionů α a β fáze jsou konstantní, koncentrace složek v mezifázové oblasti se budou postupně měnit od objemové koncentrace α na objemovou koncentraci β přes vzdálenost x. To je na rozdíl od idealizovaného Gibbsova modelu, kde vzdálenost x nabývá hodnoty nula. Diagram vpravo ukazuje rozdíly mezi skutečnými a idealizovanými modely.
Definice povrchového přebytku
V idealizovaném modelu byly chemické složky α a β hromadné fáze zůstávají nezměněny, kromě případů, kdy se blíží dělicí ploše.[3] Celkové moly jakékoli složky (příklady zahrnují: vodu, ethylenglykol atd.) Zůstávají konstantní v objemových fázích, ale liší se v povrchové fázi pro model skutečného systému, jak je uvedeno níže.
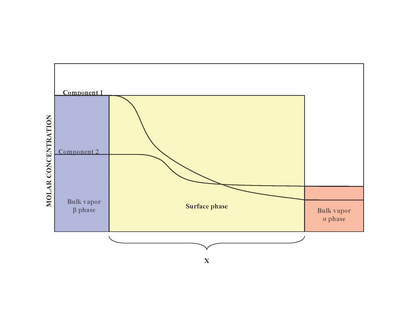
Ve skutečném systému se však celkový počet mol komponenty liší v závislosti na libovolném umístění dělícího povrchu. Kvantitativní míra adsorpce i-tá složka je zachycena povrchovým přebytečným množstvím.[1] Povrchový přebytek představuje rozdíl mezi celkovými moly i-tá složka v systému a krtci i-tá složka v konkrétní fázi (buď α nebo β) a je reprezentován:

kde Γi je povrchový přebytek i-tá složka, n jsou krtci, α a β jsou fáze a A je plocha dělícího povrchu.
Γ představuje přebytek rozpuštěné látky na jednotku plochy povrchu nad tím, co by bylo přítomno, pokud by objemová koncentrace převládala až na povrch, může být kladná, záporná nebo nulová. Má jednotky mol / m2.
Relativní přebytek povrchu
Relativní povrchové přebytečné množství jsou užitečnější než libovolné povrchové přebytečné množství.[3] Relativní povrchový přebytek souvisí s adsorpce na rozhraní s rozpouštědlem v objemové fázi. Výhodou použití relativních přebytečných množství povrchu je, že nezávisí na umístění dělícího povrchu. Relativní povrchový přebytek druhů i a rozpouštědlo 1 je tedy:

Gibbsova adsorpční izotermická rovnice
Odvození Gibbsovy adsorpční rovnice
Pro dvoufázový systém skládající se z α a β fáze v rovnováze s povrchem S dělení fází, celkem Gibbsova volná energie systému lze zapsat jako:

kde G je Gibbsova volná energie.
Rovnici Gibbsovy adsorpční izotermy lze odvodit z „partikularizace termodynamiky Eulerovy věty na homogenních formách prvního řádu“.[4] Gibbsova volná energie každé fáze α, fáze βa povrchovou fázi lze vyjádřit rovnicí:

kde U je vnitřní energie, p je tlak, PROTI je objem, T je teplota, S je entropie a μi je chemický potenciál i-tá složka.
Vezmeme-li celkovou derivaci Eulerovy formy Gibbsovy rovnice pro α fáze, β fáze a povrchová fáze:

kde A je plocha průřezu dělicí plochy a y je povrchové napětí.
U reverzibilních procesů vyžaduje první zákon termodynamiky, že:

kde q je tepelná energie a w je práce.

Dosazením výše uvedené rovnice do celkové derivace Gibbsovy energetické rovnice a využitím výsledku ydA se rovná netlakové objemové práci, když se uvažuje povrchová energie:

využitím základní rovnice Gibbsovy energie vícesložkového systému:

Rovnice vztahující se k α fáze, β fáze a povrchová fáze se stává:

Při zvažování hromadných fází (α fáze, β fáze), při rovnováze při konstantní teplotě a tlaku vyžaduje Gibbs-Duhemova rovnice, že:

Výslednou rovnicí je Gibbsova adsorpční izotermická rovnice:

Gibbsova adsorpční izoterma je rovnice, kterou lze považovat za adsorpce izoterma, která spojuje povrchové napětí roztoku s koncentrací rozpuštěné látky.
Pro binární systém obsahující dvě složky je Gibbsova adsorpční rovnice z hlediska přebytku povrchu:

Vztah mezi povrchovým napětím a povrchovou nadměrnou koncentrací
Chemický potenciál druhů i v řešení závisí na aktivitě a pomocí následující rovnice:[2]

kde μi je chemický potenciál z i-tá složka, μiÓ je chemický potenciál i-tá složka v referenčním stavu, R je plynová konstanta, T je teplota a Ai je činnost i-tá složka.
Diferenciace rovnice chemického potenciálu vede k:

kde F je koeficient aktivity složky i, a C je koncentrace druhů i v hromadné fázi.
Pokud řešení v α a β fáze jsou zředěné (bohaté na jednu konkrétní složku i) pak koeficient aktivity součásti i přistupuje k jednotě a Gibbsův izoterm se stává:

Výše uvedená rovnice předpokládá, že rozhraní je dvojrozměrné, což není vždy pravda. Tuto chybu napravují další modely, například Guggenheim.
Účinky iontové disociace
Gibbsova rovnice pro adsorpci elektrolytů
Uvažujme systém složený z vody, který obsahuje organický elektrolyt RNaz a anorganický elektrolyt NaCl, které oba disociují úplně tak, že:

Gibbsova adsorpční rovnice, pokud jde o relativní přebytek povrchu, se stává:

Vztah mezi povrchovým napětím a povrchovou nadměrnou koncentrací se stává:

kde m je koeficient Gibbsovy adsorpce.[3] Hodnoty m se počítají pomocí Dvouvrstvá (mezifázová) modely Helmholtz, Kámo, a Záď.
Látky mohou mít různé účinky na povrchové napětí, jak je znázorněno: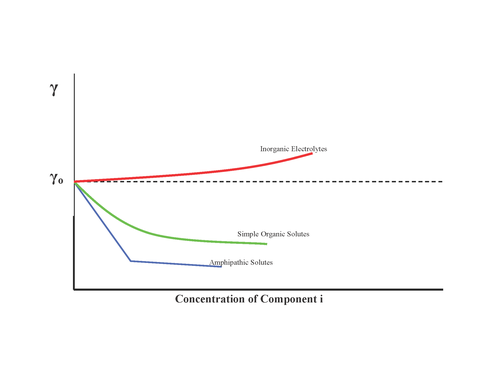
- Žádný účinek, například cukr
- Zvýšení povrchového napětí, anorganické soli
- Postupně snižujte povrchové napětí, alkoholy
- Snižte povrchové napětí a po dosažení minima již nebude mít žádný účinek: povrchově aktivní látky
Gibbsova izoterma proto předpovídá, že anorganické soli mají negativní povrchové koncentrace. Tento názor však byl v posledních letech značně zpochybněn kvůli kombinaci přesnějších mezifázově citlivých experimentů a teoretických modelů, které oba předpovídají zvýšení povrchové náchylnosti halogenidů se zvyšující se velikostí a polarizovatelností.[5] Povrchové napětí jako takové není spolehlivou metodou pro stanovení relativní náchylnosti iontů k rozhraní vzduch-voda.
K prokázání platnosti modelu je nutná metoda pro stanovení povrchových koncentrací: obvykle se používají dvě různé techniky: elipsometrie a sledování rozpadu 14C přítomný v molekulách povrchově aktivní látky.
Gibbsova izoterma pro iontové povrchově aktivní látky
Iontové povrchově aktivní látky vyžadují zvláštní úvahy elektrolyty:
- Při absenci dalších elektrolytů

kde Termín "povrchová koncentrace" označuje povrchovou koncentraci molekul povrchově aktivní látky bez ohledu na protiion.

- V přítomnosti přidaných elektrolytů


Experimentální metody
Rozsah adsorpce na kapalném rozhraní lze vyhodnotit pomocí povrchové napětí údaje o koncentraci a Gibbsova adsorpční rovnice.[3] The mikrotom Blade metoda se používá k určení hmotnosti a molal koncentrace rozhraní. Tato metoda zahrnuje dosažení jednoho metru čtverečního části rozhraní vzduch-kapalina binárních roztoků pomocí a mikrotom čepel.
Další metodou, která se používá k určení rozsahu adsorpce na rozhraní vzduch-voda, je emulzní technika, kterou lze použít k odhadu relativního povrchového přebytku vzhledem k vodě.[3]
Dále lze Gibbsův povrchový přebytek povrchově aktivní složky pro vodný roztok zjistit pomocí radioaktivní stopovač metoda. Povrchově aktivní složka je obvykle označena uhlík-14 nebo síra-35.[3]
Reference
- ^ A b C d Shchukin, E.D., Pertsov, A.V., Amelina E.A. a Zelenev, A.S. Koloidní a povrchová chemie. 1. vyd. Mobius D. a Miller R. sv. 12. Amsterdam: Elsevier Science B.V. 2001.
- ^ A b C Hiemenz, Paul C. a Rajagopalan, Raj. Principy koloidní a povrchové chemie. 3. vyd. New York: Marcel Dekker, Inc, 1997.
- ^ A b C d E F G Chattoraj, D.K. a Birdi, K.S. Adsorpce a nadbytek povrchu Gibbs. New York: Plenum Publishing Company, 1984.
- ^ Callen, Herbert B. Termodynamika a úvod do termostatiky. 2. vyd. Kanada: John Wiley & Sons, Inc, 1985.
- ^ Petersen, Poul B .; Saykally, Richard J. (2006). „O povaze iontů na povrchu kapalné vody“. Roční přehled fyzikální chemie. 57 (1): 333–364. doi:10.1146 / annurev.physchem.57.032905.104609.