Supramolekulární katalýza - Supramolecular catalysis
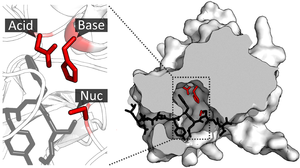
Supramolekulární katalýza není přesně definované pole, ale obecně odkazuje na aplikaci supramolekulární chemie, zejména rozpoznávání molekul a vazba hostů, na katalýzu.[1][2] Toto pole bylo původně inspirováno enzymatický systém který na rozdíl od reakcí klasické organické chemie využívá nekovalentní interakce jako je vodíková vazba, interakce kation-pi a hydrofobní síly, které dramaticky urychlují rychlost reakce a / nebo umožňují vysoce selektivní reakce. Protože enzymy jsou strukturně složité a obtížně se modifikují, supramolekulární katalyzátory nabízejí jednodušší model pro studium faktorů podílejících se na katalytické účinnosti enzymu.[3]:1 Dalším cílem, který motivuje tuto oblast, je vývoj účinných a praktických katalyzátorů, které mohou nebo nemusí mít enzymový ekvivalent v přírodě.
Úzce příbuzným studijním oborem je asymetrická katalýza který vyžaduje molekulární rozpoznávání k rozlišení dvou chirálních výchozích látek nebo chirálních přechodových stavů, a proto by mohl být kategorizován jako oblast supramolekulární katalýzy, avšak supramolekulární katalýza nemusí nutně zahrnovat asymetrickou reakci. Jak tam je další článek na Wikipedii Tento článek, který již byl napsán o asymetrických katalyzátorech s malými molekulami, se zaměřuje především na velké katalytické hostitelské molekuly. Nediskrétní a strukturálně špatně definovaný systém jako např micela a dendrimery nejsou zahrnuty.
Dějiny


Termín supramolekulární chemie definoval Jean-Marie Lehn jako „chemii intermolekulární vazby pokrývající struktury a funkce entit vytvořených asociací dvou nebo více chemických druhů“ ve své Nobelově přednášce v roce 1987,[6] ale koncept supramolekulární katalýzy začal již dříve v roce 1946 Linus Pauling, když založil teorii enzymové katalýzy, při které je zrychlení rychlosti výsledkem nekovalentní stabilizace přechodového stavu pomocí enzymů.[7] Nicméně až o několik desetiletí později byl vyvinut umělý enzym. První jednoduché napodobeniny enzymů byly založeny na korunovém etheru a kryptandě.[8] V roce 1976, necelých deset let po objevení korunového etheru, Cram et al. vyvinuli funkcionalizovaný binaftylkorunový ether, který katalyzuje transacylaci.[4] Katalyzátor využívá schopnost motivu korunového etheru zachytit kation k navázání na amoniovou iontovou část substrátu a následně využívá blízký thiolový motiv ke štěpení esteru.
Od začátku 70. let cyklodextriny byly rozsáhle studovány pro své enkapsulační vlastnosti a použity jako vazebná místa v supramolekulárním katalyzátoru.[2] Cyklodextriny mají tuhou prstencovou strukturu, hydrofilní povrch a hydrofobní dutinu uvnitř; proto jsou schopné vázat organické molekuly ve vodném roztoku. V roce 1978, se základními znalostmi, že hydrolýza m-terc-butylfenylacetátu je urychlena v přítomnosti 2-benzimidazol-octové kyseliny a alfa-cyklodextrinu,[9] Brewslow a kol. vyvinul katalyzátor na bázi beta-cyklodextrinu nesoucího dvě imidazolové skupiny. Tento cyklodextrinový katalytický systém napodobuje ribonukleázu A tím, že používá neutrální imidazol a imidazolový kation pro selektivní štěpení cyklických fosfátových substrátů. Rychlost reakce je katalyzována 120krát rychleji a na rozdíl od hydrolýzy jednoduchou bází NaOH, která poskytuje směs produktů 1: 1, poskytují tyto katalyzátory selektivitu 99: 1 pro jednu sloučeninu.[5]
V roce 1993 Rebek a kol. vyvinuli první samostatně sestavitelnou kapsli[10] a v roce 1997 byla pro katalyzování Diels-Alderovy reakce použita takzvaná struktura „tenisového míčku“.[11] Samostatně sestavené molekuly mají oproti korunovému etheru a cyklodextrinu výhodu v tom, že mohou zachytit významné větší molekuly nebo dokonce dvě molekuly najednou. V následujících desetiletích mnoho výzkumných skupin, jako je Makoto Fujita, Ken Raymond, a Jonathan Nitschke, vyvinuli katalyzátory podobné kleci také z molekulární samo-sestavení zásada.
V roce 2002 Sanders a spolupracovníci publikovali použití techniky dynamické kombinatorické knihovny ke konstrukci receptoru[12] a v roce 2003 použili techniku k vývoji katalyzátoru pro Diels-Alderovu reakci.[13]
Mechanismus katalýzy
Zde jsou popsány tři běžné způsoby katalýzy.
Orientace reaktivních a labilních skupin

Supramolekulární hostitel se může vázat na molekulu hosta takovým způsobem, že labilní skupina hosta je umístěna blízko reaktivní skupiny hostitele. Blízkost těchto dvou skupin zvyšuje pravděpodobnost, že by mohlo dojít k reakci, a tím se zvýší rychlost reakce. Tento koncept je podobný principu preorganizace který uvádí, že komplexace by mohla být vylepšena, pokud jsou vazebné motivy preorganizované v dobře definované poloze, takže hostitel nevyžaduje pro komplexaci žádnou zásadní konformační změnu.[15] V tomto případě je katalyzátor předorganizován tak, že nejsou nutné žádné velké konformační změny, aby reakce proběhla. Pozoruhodným příkladem katalyzátorů, které používají tento mechanismus, je korunní ether Jean-Marie Lehna.[14] Kromě toho katalyzátory na bázi funkcionalizovaných cyklodextrinů často využívají tento způsob katalýzy.[16]:88
Zvýšení účinné koncentrace substrátu
Bimolekulární reakce jsou vysoce závislé na koncentraci substrátů. Proto, když supramolekulární nádoba zapouzdří oba reaktanty ve své malé dutině, efektivní lokální koncentrace reakčních složek se zvyšuje a v důsledku entropického účinku se zrychluje rychlost reakce.[16]:89 To znamená, že intramolekulární reakce je rychlejší než její odpovídající intermolekulární reakce.
I když je pozorován vysoký vzestup účinné koncentrace, molekuly, které používají tento způsob katalýzy, mají ve srovnání s enzymy malé zrychlení rychlosti. Navrhované vysvětlení je, že v nádobě nejsou substráty tak pevně vázány jako v enzymu. Reagencie mají v dutině prostor kroutí se, takže entropický účinek nemusí být tak důležitý. Dokonce i v případě enzymů výpočtové studie ukázaly, že entropický účinek může být také nadhodnocen.[17]
Příklady molekul, které fungují prostřednictvím tohoto mechanismu, jsou Rebekův tenisový míček a Fujitaův oktaedrický komplex.[11][18]

Stabilizační přechodový stav
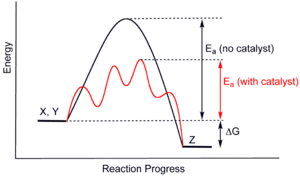
Supramolekulární katalyzátory mohou urychlit reakce nejen umístěním dvou reaktantů do těsné blízkosti, ale také stabilizací přechodového stavu reakce a snížením aktivační energie.[16]:89 Zatímco tohle základní princip katalýzy je běžný u malých molekul nebo heterogenních katalyzátorů, supramolekulární katalyzátory však mají obtížné využití konceptu kvůli jejich často tuhým strukturám. Na rozdíl od enzymů, které mohou měnit tvar, aby se přizpůsobily substrátům, supramolekuly nemají tento druh flexibility a tak zřídka dosahují úpravy sub-angstromu potřebné pro perfektní stabilizaci přechodového stavu.[3]:2
Příkladem katalyzátorů tohoto typu je Sanderův porfyrinový trimer. Diels Alderova reakce mezi dvěma substráty funkcionalizovanými pyridinem obvykle vede ke směsi produktů endo a exo. V přítomnosti dvou katalyzátorů však bylo možné dosáhnout úplné endo selektivity nebo exo selektivity. Základní příčinou selektivity je koordinační interakce mezi pyridinem a iontem zinku na porfyrinu. V závislosti na tvaru katalyzátorů je upřednostňován jeden produkt před druhým.[19]

Přístupy k výrobě supramolekulárních katalyzátorů
Designový přístup
Tradiční přístup k supramolekulárním katalyzátorům se zaměřuje na návrh makromolekulárního receptoru s vhodně umístěnými katalytickými funkčními skupinami. Tyto katalyzátory jsou často inspirovány strukturou enzymů s katalytickou skupinou napodobující reaktivní aminokyselinové zbytky, ale na rozdíl od skutečných enzymů jsou vazebná místa těchto katalyzátorů tuhá struktura vyrobená z chemických stavebních bloků.[20] Všechny příklady v tomto článku jsou vyvinuty pomocí návrhového přístupu.
Jeremy Sanders poukázal na to, že designový přístup nebyl úspěšný a kvůli tuhosti supramolekul vyrobil velmi málo účinných katalyzátorů. Tvrdil, že tuhé molekuly s mírným nesouladem s přechodovým stavem nemohou být účinným katalyzátorem. Spíše než investovat tolik úsilí do jedné tuhé molekuly, že nemůžeme určit její přesnou geometrii na úroveň sub-angstromu, která je nutná pro dobrou stabilizaci, navrhl Sanders použití mnoha malých flexibilních stavebních bloků s konkurenčními slabými interakcemi, aby bylo možné aby katalyzátor upravil svou strukturu tak, aby lépe vyhovoval podkladu.[21] Existuje přímý kompromis mezi entalpickým přínosem z flexibilní struktury a entropickým přínosem z tuhé struktury.[3]:3 Flexibilní struktura by snad mohla lépe vázat přechodový stav, ale poskytuje více prostoru pro pohyb a vibrace substrátů. Většina supramolekulárních chemiků v minulosti raději stavěla rigidní struktury ze strachu z entropických nákladů.[21]
Tento problém by snad mohl napravit Pekař a Houk „přístup naruby“, který umožňuje systematický vývoj enzymů de novo.[22] Tato výpočetní metoda začíná jednoduše předpovězenou strukturou přechodového stavu a pomalu se staví směrem ven optimalizací uspořádání funkčních skupin ke stabilizaci přechodového stavu. Poté vyplní zbytek aktivního místa a nakonec generuje celé proteinové lešení, které by mohlo obsahovat navržené aktivní místo. Tuto metodu lze potenciálně použít na supramolekulární katalýzu, i když nepřeberné množství chemických stavebních bloků by mohlo snadno přemoci výpočetní model určený pro práci s 20 aminokyselinami.
Přechodový stav analogového výběru / screeningu
Za předpokladu, že katalytická aktivita do značné míry závisí na afinitě katalyzátoru k přechodnému stavu, lze syntetizovat a přechodový stav analogový (TSA), struktura, která se podobá přechodovému stavu reakce. Pak by se dalo spojit TSA s pevným nosičem nebo identifikovatelnou značkou a použít tento TSA k výběru optimálního katalyzátoru ze směsi mnoha různých potenciálních katalyzátorů generovaných chemicky nebo biologicky syntéza orientovaná na rozmanitost. Tato metoda umožňuje rychlý screening knihovny různých sloučenin. Nevyžaduje tolik syntetického úsilí a umožňuje studium různých katalytických faktorů současně. Metoda by tedy mohla potenciálně přinést účinný katalyzátor, který bychom podle našich současných znalostí nemohli navrhnout.[20]
Mnoho katalytické protilátky byly vyvinuty a studovány pomocí tohoto přístupu.
Přístup skríningu katalytické aktivity

Problém s přístupem k výběru analogového přechodu je, že katalytická aktivita není screeningovým kritériem. TSA nemusí nutně představovat skutečné přechodové stavy, a tak katalyzátor získaný skríninkem může být jen nejlepším receptorem pro TSA, ale nemusí být nutně nejlepším katalyzátorem. Aby se tento problém obešel, je třeba měřit katalytickou aktivitu přímo a také rychle. Vyvinout a vysoce výkonná obrazovka, substráty mohou být navrženy tak, aby po reakci změnily barvu nebo uvolnily fluorescenční produkt. Například Crabtree a spolupracovníci využili tuto metodu při screeningu hydrosylačních katalyzátorů na alken a imin.[23] Předpokladem pro takové substráty je bohužel zúžení rozsahu reakcí pro studium.[20]
Přístup dynamické kombinatorické knihovny

Na rozdíl od tradiční kombinatorické syntézy, kde byla nejprve vytvořena knihovna katalyzátorů a později prověřována (jako ve dvou výše uvedených přístupech), dynamická kombinatorická knihovna Tento přístup využívá směs vícesložkových stavebních bloků, které reverzibilně tvoří knihovnu katalyzátorů. Bez šablony se knihovna skládá ze zhruba stejné směsi různých kombinací stavebních bloků. V přítomnosti šablony, která je buď výchozím materiálem nebo TSA, je kombinace, která poskytuje nejlepší vazbu na šablonu, termodynamicky příznivá, a proto je tato kombinace častější než ostatní členové knihovny. Předpjatý poměr požadovaného katalyzátoru k jiným kombinatorickým produktům by pak mohl být zmrazen ukončením reverzibility rovnováhy pomocí změn teploty, pH nebo záření, čímž se získá optimální katalyzátor.[20] Například Lehn et al. použil tuto metodu k vytvoření dynamické kombinatorické knihovny iminového inhibitoru ze sady aminů a sady aldehydů. Po nějaké době byla rovnováha ukončena přidáním NaBH3CN, čímž se získá požadovaný katalyzátor.[24]
Prominentní příklady supramolekulárních katalyzátorů
Diederichova napodobenina pyruvát oxidázy
V přírodě, pyruvát oxidáza zaměstnává dva kofaktory thiamin pyrofosfát (ThDP) a Flavin adenin dinukleotid (FAD) ke katalýze přeměny pyruvátu na acetylfosfát. Nejprve ThDP zprostředkovává dekarboxylaci pyruvátu a generuje aktivní aldehyd jako produkt. Aldehyd je poté oxidován FAD a následně je atakován fosfátem za vzniku acetylfosfátu.
Diederich a spolupracovníci napodobili tento systém supramolekulárním katalyzátorem založeným na cyklofan. Katalyzátor má thiazoliový ion, reaktivní část ThDP a flavin, jádro FAD s holými kostmi, v těsné blízkosti a v blízkosti místa vázajícího substrát. Katalytický cyklus je téměř stejný jako v přírodě, kromě toho, že substrátem je spíše aromatický aldehyd než pyruvát. Nejprve se katalyzátor váže na substrát ve svém cyklofanovém kruhu. Poté používá thiazoliový ion ke kondenzaci se substrátem za vzniku aktivního aldehydu. Tento aldehyd je oxidován flavinem a poté napaden methanolem, čímž se získá methylester.[25]

Nolteův postupný epoxidační katalyzátor pro alkenový polymer
Procesní enzymy jsou proteiny, které katalyzují po sobě jdoucí reakce bez uvolnění substrátu. Příkladem procesních enzymů je RNA polymeráza, která se váže na řetězec DNA a opakovaně katalyzuje přenosy nukleotidů, čímž účinně syntetizuje odpovídající řetězec RNA.
Společnost Nolte a spolupracovníci vyvinuli umělý procesní enzym ve formě manganového porfyrinového rotaxanu, který šlape podél dlouhého polymeru alkenu a katalyzuje několik kol epoxidace alkenu. Ion manganatý v porfyrinu je katalytické centrum molekuly, schopné epoxidace v přítomnosti donoru kyslíku a aktivačního ligandu. S malým ligandem, jako je pyridin, který váže mangan zevnitř dutiny rotaxanu, dochází k epoxidaci mimo katalyzátor. U velkého objemného ligandu, jako je terc-butylpyridin, který se nevejde do dutiny, však dochází k epoxidaci na vnitřní straně katalyzátoru.[26]

Raymondův Nazarovův cyklický katalyzátor
Raymond a spolupracovníci vyvinuli supramolekulárního hostitele M.4L6 (4 ionty gália a 6 ligandů pro každý komplex), které se samy shromažďují prostřednictvím interakce kov-ligand ve vodném roztoku. Tato molekula zásobníku je polyaniontová, a proto je její dutina ve tvaru čtyřstěnu schopná zapouzdřit a stabilizovat kationtovou molekulu. V důsledku toho lze enkapsulovanou molekulu snadno protonovat, protože výsledná karbokace z protonace je stabilizována okolními anionty. Raymond využil tuto vlastnost k provedení kyselinou katalyzované Nazarovovy cyklizace. Katalyzátor urychluje reakci více než milionkrát, což z něj činí dosud nejúčinnější supramolekulární katalyzátor. Bylo navrženo, že taková vysoká katalytická aktivita nevyplývá pouze ze zvýšené bazicity zapouzdřeného substrátu, ale také z konstriktivní vazby, která stabilizuje přechodový stav cyklizace. Tento katalyzátor má bohužel problém inhibice produktu. K překonání tohoto problému by produkt cyklizační reakce mohl reagovat s dienofilem, který jej transformuje na a Diels-Alder adukt, který se už nevejde do dutiny katalyzátoru.[1]
V tomto případě byl supramolekulární hostitel původně navržen tak, aby jednoduše zachytil kationtové hosty. Téměř o deset let později byl využíván jako katalyzátor Nazarovovy cyklizace.

Chirálně chirálně sestavený katalyzátor pro asymetrické [2 + 2] fotoadice
Fujita a spolupracovníci objevili samo-sestavitelnou M6L4 (6 iontů palladia a 4 ligandy v každém komplexu) supramolekulární nádoba, která by mohla být vylepšena na chirální supramolekula přidáním periferní chirální pomocné látky. V tomto případě pomocný diethyldiaminocyklohexan neaktivuje přímo katalytické místo, ale vyvolává mírnou deformaci triazinové roviny za vzniku chirální dutiny uvnitř molekuly zásobníku. Tento kontejner by pak mohl být použit pro asymetrickou katalýzu [2 + 2] fotoadice maleimidu a inertní aromatické sloučeniny fluoranthenu, u kterých dosud nebylo prokázáno, že by procházely termickou nebo fotochemickou pericyklickou reakcí. Katalyzátor poskytuje enantiomerní přebytek 40%.[27]

Listova omezená Bronstedova kyselina jako katalyzátor pro asymetrickou spiroacetalizaci
Seznam a spolupracovníci, inspirovaní enzymy s hlubokou kapsou aktivního místa, navrhli a zkonstruovali sadu omezených Bronstedových kyselin s extrémně stericky náročnou chirální kapsou založenou na C2-symetrická kyselina bis (binaftyl) imidodifosforečná. V chirálním mikroprostředí mají katalyzátory geometricky fixované bifunkční aktivní místo, které aktivuje jak elektrofilní část, tak nukleofilní část substrátu. Tento katalyzátor umožňuje stereoselektivní tvorbu spiroacetalu s vysokým enantiomerním přebytkem pro různé substráty.[28]

Supramolekulární inhibitory
Supramolekulární zásobníky nemají uplatnění pouze při katalýze, ale také v opačném smyslu, konkrétně v inhibici. Kontejnerová molekula by mohla zapouzdřit hostující molekulu a následně by hosta učinila nereaktivním. Mechanismus inhibice může být buď to, že substrát je zcela izolován od činidla, nebo že molekula zásobníku destabilizuje přechodový stav reakce.
Nitschke a spolupracovníci vynalezli samo-montáž M4L6 supramolekulární hostitel s čtyřboká hydrofobní dutina, která se může zapouzdřit bílý fosfor. Samozápalný fosfor, který by se při kontaktu se vzduchem mohl samovolně vznítit, je v dutině vzduchově stabilní. I když je otvor v dutině dostatečně velký, aby do něj mohla vstoupit molekula kyslíku, přechodový stav spalování je příliš velký, aby se vešel do dutiny malé klece.[29]

Problémy a omezení
Po mnoha desetiletích od svého vzniku zůstává aplikace supramolekulární chemie v praktické katalýze nepolapitelná. Supramolekulární katalýza dosud významně nepřispěla v oblasti průmyslové chemie nebo syntetické metodologie.[21] Zde je několik problémů spojených s tímto polem.
Inhibice produktu
V mnoha supramolekulárních katalytických systémech navržených pro práci s bimolekulárními adičními reakcemi, jako je Diels-Alder, se produkt reakce váže na supramolekulárního hostitele silněji než dva substráty, což vede k inhibici produktem. Výsledkem je, že tyto katalyzátory mají číslo obratu jedna a nejsou skutečně katalytické. Pro úplnou konverzi je zapotřebí stechiometrické množství katalyzátorů.[30]
Špatná stabilizace přechodového stavu
Většina supramolekulárních katalyzátorů se vyvíjí z tuhých stavebních bloků, protože tuhé bloky jsou při konstrukci požadovaného tvaru a umístění funkčních skupin tam, kam chce návrhář, méně komplikované než pružné části. Kvůli tuhosti však mírný nesoulad od přechodového stavu nevyhnutelně vede ke špatné stabilizaci a tím ke špatné katalýze. V přírodě jsou enzymy pružné a mohly by změnit své struktury tak, aby vážily přechodový stav lépe než jejich nativní forma.[21]
Obtíž při syntéze a další úpravě
Syntézy velkých komplexních katalyzátorů jsou náročné na čas a zdroje. Neočekávaná odchylka od návrhu by mohla být katastrofální. Jakmile je katalyzátor objeven, může být modifikace pro další nastavení tak synteticky náročná, že je snazší studovat špatný katalyzátor než ho vylepšit.[21]
Viz také
Reference
- ^ A b Raymond, K. N.; Hastings, C. J .; Pluth, M. D .; Bergman, R. G. (2010). „Enzymelike Catalysis of the Nazarov Cyclization by Supramolecular Encapsulation“. Journal of the American Chemical Society. 132 (20): 6938–6940. doi:10.1021 / ja102633e. PMID 20443566.
- ^ A b Nolte, R. J. M .; Vriezema, D. M .; Aragone, M. C .; Elemans, J. J. A. W .; Cornelissen, J. J. L. M .; Rowan, A. E. (2005). "Samonosné nanoreaktory". Chemické recenze. 105 (4): 1445–1489. doi:10.1021 / cr0300688. hdl:2066/32981. PMID 15826017.
- ^ A b C van Leeuwen, P. W. N. M. (2008). Supramolekulární katalýza. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA. ISBN 978-3-527-32191-9.
- ^ A b Cram, D. J .; Chao, Y. (1976). "Enzymové mechanismy, modely a napodobeniny". Journal of the American Chemical Society. 98 (4): 1015–1017. doi:10.1021 / ja00420a026.
- ^ A b Breslow, R .; Doherty, J. B .; Guillot, G .; Lipsey, C. (1978). "Použití cykloamylosy k testování systému nabíjecího relé". Journal of the American Chemical Society. 100 (10): 3227–3229. doi:10.1021 / ja00478a052.
- ^ Lehn, J. (1988). "Výběr a zesílení katalyzátoru z dynamické kombinatorické knihovny". Angewandte Chemie International Edition. 27 (1): 89–112. doi:10,1002 / anie.198800891.
- ^ Pauling, L. (1946). „Molekulární architektura a biologické reakce“ (PDF). Chemické a technické novinky. 24 (10): 1375–1377. doi:10.1021 / cen-v024n010.p1375.
- ^ Kirby, A. J. (1996). "Enzymové mechanismy, modely a napodobeniny". Angewandte Chemie International Edition. 35 (7): 706–724. doi:10.1002 / anie.199607061.
- ^ Bender, M.L .; Komiyama, M .; Breaux, E. J. (1977). "Použití cykloamylosy k testování systému nabíjecího relé". Bioorganická chemie. 6 (2): 127–136. doi:10.1016/0045-2068(77)90015-3.
- ^ Rebek, J. Jr.; Wyler, R .; de Mendoza J. (1993). „Syntetická dutina se shromažďuje prostřednictvím komplementárních vodíkových vazeb“. Angewandte Chemie International Edition. 32 (12): 1699–1701. doi:10.1002 / anie.199316991.
- ^ A b Rebek, J. ml; Kang, J. (1997). „Zrychlení reakce Diels – Alder pomocí samo-sestavené molekulární kapsle“. Příroda. 385 (661): 50–52. Bibcode:1997 Natur.385 ... 50K. doi:10.1038 / 385050a0. PMID 8985245.
- ^ Sanders, J. K. M .; Otto, S .; Furlan, R. L. E. (2002). "Výběr a zesílení hostitelů z dynamických kombinatorických knihoven makrocyklických disulfidů". Věda. 297 (5581): 590–593. Bibcode:2002Sci ... 297..590O. doi:10.1126 / science.1072361. PMID 12142534.
- ^ Otto, S .; Brisig, B .; Sanders, J. K. M. (2003). "Výběr a zesílení katalyzátoru z dynamické kombinatorické knihovny". Angewandte Chemie International Edition. 42 (11): 1270–1273. doi:10.1002 / anie.200390326. PMID 12645061.
- ^ A b Lehn, J.; Sirlin, C. (1978). „Molecular Catalysis: Enhanced Rates of Thiolysis with High Structural and Chiral Recognition in Complexes of Reactive Macrocyclic Receptor Molecule“. Chemická komunikace (21): 949–951. doi:10.1039 / C39780000949.
- ^ Cram, D. J. (1988). „Návrh molekulárních hostitelů, hostů a jejich komplexů“. Angewandte Chemie International Edition. 27 (8): 1009–1020. doi:10,1002 / anie.198810093.
- ^ A b C Beer, P .; Gale, P. A .; Smith, D. K. (1999). Supramolekulární chemie. New York: Oxford University Press. ISBN 978-0-19-850447-4.
- ^ Warshel, A .; Aaqvist, J. (1993). „Návrh molekulárních hostitelů, hostů a jejich komplexů“. Chemické recenze. 93 (7): 2523–2544. doi:10.1021 / cr00023a010.
- ^ Fujita, M .; Yoshizawa, M .; Tamura, M. (2006). „Diels-Alder ve vodných molekulárních hostitelích: neobvyklá regioselektivita a účinná katalýza“. Věda. 312 (5771): 251–254. Bibcode:2006Sci ... 312..251Y. doi:10.1126 / science.1124985. PMID 16614218.
- ^ Sanders, J. K. M.; Walter, C. J .; Anderson, H. L. (1993). „Exo-selektivní zrychlení intermolekulární reakce Diels-Alder podle hostitele trimerního porfyrinu“. Chemická komunikace (5): 458–460. doi:10.1039 / C39930000458.
- ^ A b C d Motherwell, W. B .; Bingham, M. J .; Šest, Y. (2001). "Nedávný pokrok v designu a syntéze umělých enzymů". Čtyřstěn. 57 (22): 4663–4686. doi:10.1016 / S0040-4020 (01) 00288-5.
- ^ A b C d E Sanders, J. K. M. (1998). "Supramolekulární katalýza v přechodu". Chemistry: A European Journal. 4 (8): 1378–1383. doi:10.1002 / (SICI) 1521-3765 (19980807) 4: 8 <1378 :: AID-CHEM1378> 3.0.CO; 2-3.
- ^ Houk, K. N .; Kiss, G .; Çelebi-Ölçüm, N .; Moretti, R .; Baker, D. (2013). "Výpočetní enzymový design". Angewandte Chemie International Edition. 52 (22): 5700–5725. doi:10,1002 / anie.201204077.
- ^ Crabtree, R. H.; Cooper, A. C .; McAlexander, L. H .; Lee, D.-H .; Torres, M. T. (1998). „Reaktivní barviva jako metoda rychlého screeningu homogenních katalyzátorů“. Journal of the American Chemical Society. 120 (38): 9971–9972. doi:10.1021 / ja9818607.
- ^ Lehn, J.; Huc, I. (1997). „Virtuální kombinatorické knihovny: dynamické generování molekulární a supramolekulární rozmanitosti samoobsluhou“. PNAS. 94 (6): 2106–2110. Bibcode:1997PNAS ... 94.2106H. doi:10.1073 / pnas.94.6.2106. PMC 20048. PMID 9122156.
- ^ Diederich, F .; Mattei, P. (1997). "Catalytic Cyclophanes. Part XI. A Flavo-thiazolio-cyklophane as a Biomimetic Catalyst for the Preparative-scale Electro-oxidation of Aromatic Aldehydes to Methyl Esters". Helvetica Chimica Acta. 80 (5): 1555–1588. doi:10.1002 / hlca.19970800516.
- ^ Nolte, R. J. M .; Thordarson, P .; Bijsterveld, E. J. A .; Rowan, A. E. (2003). "Epoxidace polybutadienu topologicky vázaným katalyzátorem". Příroda. 424 (6951): 915–918. Bibcode:2003 Natur.424..915T. doi:10.1038 / nature01925. PMID 12931181.
- ^ Fujita, M .; Nishioka, Y .; Yamaguchi, T .; Kawano, M. (2008). „Asymetrická [2 + 2] olefinová křížová fotoadice v samo-sestaveném hostiteli se vzdálenými chirálními pomocnými prostředky“. Journal of the American Chemical Society. 130 (26): 8160–8161. doi:10.1021 / ja802818t. PMID 18540605.
- ^ List, B .; Čorić, I. (2012). „Asymetrická spiroacetalizace katalyzovaná uzavřenými Brønstedovými kyselinami“. Příroda. 483 (7389): 315–319. Bibcode:2012Natur.483..315C. doi:10.1038 / příroda10932. PMID 22422266.
- ^ Nitschke, J. R .; Mal, P .; Breiner, B .; Rissanen, K. (2009). „Bílý fosfor je vzduchem stabilní v samo-sestavené čtyřboké tobolce“. Věda. 324 (5935): 1697–1699. Bibcode:2009Sci ... 324.1697M. doi:10.1126 / science.1175313. PMID 19556504.
- ^ Easton, C. J .; Lincoln, S. F .; Barr, L .; Onagi H. (2004). "Molekulární reaktory a stroje: aplikace, potenciál a omezení". Chemistry: A European Journal. 10 (13): 3120–3128. doi:10.1002 / chem.200305768. PMID 15224320.