Bloomovy filtry v bioinformatice - Bloom filters in bioinformatics
Bloom filtry jsou prostorově efektivní pravděpodobnostní datové struktury slouží k otestování, zda je prvek součástí sady. Bloom filtry vyžadují mnohem méně prostoru než jiné datové struktury pro reprezentaci sad, avšak nevýhodou Bloomových filtrů je, že existuje míra falešně pozitivních výsledků při dotazování na datovou strukturu. Vzhledem k tomu, že několik prvků může mít stejné hodnoty hash pro řadu hash funkcí, je pravděpodobné, že dotazování na neexistující prvek může vrátit klad, pokud byl do filtru Bloom přidán jiný prvek se stejnými hodnotami hash. Za předpokladu, že hashovací funkce má stejnou pravděpodobnost výběru libovolného indexu Bloomova filtru, je falešná kladná rychlost dotazování Bloomova filtru funkcí počtu bitů, počtu hashovacích funkcí a počtu prvků Bloomova filtru. To umožňuje uživateli řídit riziko získání falešně pozitivního výsledku kompromitováním prostorových výhod filtru Bloom.
Bloom filtry se primárně používají v bioinformatice k testování existence a k-mer v sekvenci nebo sadě sekvencí. K-mery sekvence jsou indexovány v Bloom filtru a jakýkoli k-mer stejné velikosti lze dotazovat proti Bloom filtru. Toto je výhodnější alternativa k hashování k-mery sekvence s a hash tabulka, zvláště když je sekvence velmi dlouhá, protože je velmi náročné ukládat do paměti velké množství k-merů.
Aplikace
Charakterizace sekvence
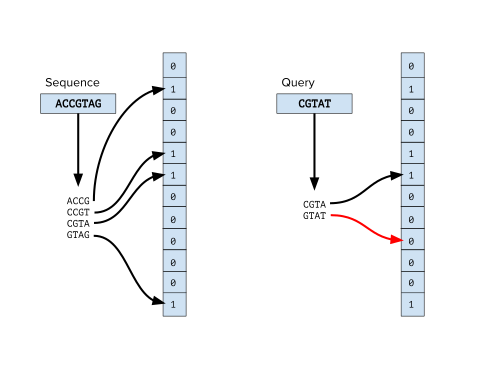
Krok předzpracování v mnoha bioinformatických aplikacích zahrnuje klasifikaci sekvencí, primárně klasifikaci čtení z a Sekvenování DNA experiment. Například v metagenomické studie je důležité vědět, zda sekvenční čtení patří novému druhu.[1] a v projektech klinického sekvenování je životně důležité odfiltrovat čtení z genomů kontaminujících organismů. Existuje mnoho nástrojů pro bioinformatiku, které používají Bloomovy filtry ke klasifikaci čtení dotazováním k-merů čtení na sadu Bloomových filtrů generovaných ze známých referenční genomy. Některé nástroje, které používají tuto metodu, jsou FACS[2] a nástroje BioBloom.[3] I když tyto metody nemusí překonat jiné nástroje pro klasifikaci bioinformatiky, jako je Kraken,[4] nabízejí alternativu efektivní z hlediska paměti.
Nedávná oblast výzkumu s Bloomovými filtry v charakterizaci sekvence je ve vývoji způsobů dotazování surových čtení ze sekvenčních experimentů. Například, jak lze určit, která čtení obsahují konkrétní 30mer v celém NCBI Archiv čtení sekvence ? Tento úkol je podobný tomu, kterého je dosaženo pomocí VÝBUCH, nicméně zahrnuje dotazování mnohem větší datové sady; zatímco BLAST dotazy na databázi referenčních genomů, tento úkol vyžaduje vrácení konkrétních čtení, která obsahují k-mer. BLAST a podobné nástroje nemohou tento problém efektivně zvládnout, proto byly za tímto účelem implementovány datové struktury založené na Bloom filtru. Binární květinové stromy[5] jsou binární stromy Bloomových filtrů, které usnadňují dotazování na přepisy ve velkém RNA sekvence experimenty. VELKÉ[6] půjčuje si bitcovité podpisy z oblasti načítání dokumentů indexovat a dotazovat se na veškerá data o mikrobiálním a virovém sekvenování v souboru Evropský archiv nukleotidů. Podpis dané datové sady je zakódován jako sada Bloomových filtrů z této datové sady.
Sestavení genomu

Účinnost paměti filtrů Bloom byla použita v shromáždění genomu jako způsob, jak snížit prostorovou stopu k-merů ze sekvenování dat. Přínosem montážních metod založených na Bloomových filtrech je kombinace Bloomových filtrů a de Bruijn grafy do struktury zvané pravděpodobnostní de Bruijnův graf,[7] který optimalizuje využití paměti za cenu falešně pozitivních hodnot inherentních filtrům Bloom. Namísto uložení de Bruijnova grafu v hašovací tabulce je uložen ve filtru Bloom.
Použití Bloomova filtru k uložení de Bruijnova grafu komplikuje krok přechodu grafu k sestavení sestavy, protože informace o okraji nejsou ve Bloomově filtru zakódovány. Traversal grafu se provádí dotazem na Bloomův filtr pro kterýkoli ze čtyř možných následných k-merů z aktuálního uzlu. Například pokud je aktuální uzel pro k-mer ACT, pak další uzel musí být pro jeden z k-mer CTA, CTG, CTC nebo CTT. Pokud ve filtru Bloom existuje k-mer dotazu, pak se k cestě přidá k-mer. Proto existují dva zdroje falešných poplachů při dotazování Bloomova filtru při procházení de Bruijnova grafu. Existuje pravděpodobnost, že jeden nebo více ze tří falešných k-merů existuje jinde v sadě sekvencování pro vrácení falešně pozitivního, a existuje výše uvedená inherentní míra falešných pozitivů samotného filtru Bloom. Montážní nástroje, které používají Bloomovy filtry, musí ve svých metodách tyto zdroje falešných poplachů zohledňovat. ABySS 2[8] a Minia[9] jsou příklady assemblerů, které tento přístup používají de novo shromáždění.
Oprava chyby sekvenování
Sekvenování nové generace Metody (NGS) umožnily generování nových sekvencí genomu mnohem rychleji a levněji než předchozí Sangerovo sekvenování metody. Tyto metody však mají vyšší chybovost,[10][11] což komplikuje následnou analýzu sekvence a může dokonce vést k chybným závěrům. Bylo vyvinuto mnoho metod k opravě chyb ve čtení NGS, ale využívají velké množství paměti, což je činí nepraktickými pro velké genomy, jako je lidský genom. Proto byly vyvinuty nástroje využívající Bloom filtry k řešení těchto omezení, využívajících výhod jejich efektivního využití paměti. Mušketa[12] a BLESS[13] jsou příklady takových nástrojů. Obě metody používají pro korekci chyb přístup k-merního spektra. Prvním krokem tohoto přístupu je spočítat multiplicitu k-merů, avšak zatímco BLESS používá k ukládání počtů pouze Bloomovy filtry, Musket používá Bloomovy filtry pouze k počítání jedinečných k-merů a ne-jedinečné k-mery ukládá do hash tabulka, jak je popsáno v předchozí práci[14]
RNA-sekv
V některých jsou také použity filtry Bloom RNA-sekv potrubí. RNA-Skim[15] shluky RNA přepisy a poté pomocí Bloomových filtrů najde sig-méry: k-mery, které se nacházejí pouze v jednom z klastrů. Tyto signály se poté použijí k odhadu úrovní hojnosti přepisu. Proto neanalyzuje všechny možné k-mer, které vedou ke zlepšení výkonu a využití paměti, a ukázalo se, že funguje stejně jako předchozí metody.
Reference
- ^ Lundeberg, Joakim; Arvestad, Lars; Andersson, Björn; Allander, Tobias; Käller, Max; Stranneheim, Henrik (01.07.2010). "Klasifikace sekvencí DNA pomocí Bloomových filtrů". Bioinformatika. 26 (13): 1595–1600. doi:10.1093 / bioinformatika / btq230. ISSN 1367-4803. PMC 2887045. PMID 20472541.
- ^ Lundeberg, Joakim; Arvestad, Lars; Andersson, Björn; Allander, Tobias; Käller, Max; Stranneheim, Henrik (01.07.2010). "Klasifikace sekvencí DNA pomocí Bloomových filtrů". Bioinformatika. 26 (13): 1595–1600. doi:10.1093 / bioinformatika / btq230. ISSN 1367-4803. PMC 2887045. PMID 20472541.
- ^ Chu, Justin; Sadeghi, Sara; Raymond, Anthony; Jackman, Shaun D .; Nip, Ka Ming; Mar, Richard; Mohamadi, Hamid; Butterfield, Yaron S .; Robertson, A. Gordon (01.12.2014). „Nástroje BioBloom: rychlý, přesný a paměťově efektivní screening sekvence hostitelských druhů pomocí Bloom filtrů“. Bioinformatika. 30 (23): 3402–3404. doi:10.1093 / bioinformatika / btu558. ISSN 1367-4811. PMC 4816029. PMID 25143290.
- ^ Wood, Derrick E .; Salzberg, Steven L. (03.03.2014). „Kraken: ultrarychlá klasifikace metagenomických sekvencí pomocí přesných zarovnání“. Genome Biology. 15 (3): R46. doi:10.1186 / gb-2014-15-3-r46. ISSN 1474-760X. PMC 4053813. PMID 24580807.
- ^ Carl Kingsford; Solomon, Brad (březen 2016). „Rychlé hledání tisíců krátkých sekvenčních experimentů“. Přírodní biotechnologie. 34 (3): 300–302. doi:10,1038 / nbt.3442. ISSN 1546-1696. PMC 4804353. PMID 26854477.
- ^ Iqbal, Zamin; McVean, Gil; Rocha, Eduardo P. C .; Bakker, Henk C. den; Bradley, Phelim (únor 2019). „Ultrarychlé vyhledávání všech uložených bakteriálních a virových genomových dat“. Přírodní biotechnologie. 37 (2): 152–159. doi:10.1038 / s41587-018-0010-1. ISSN 1546-1696. PMC 6420049. PMID 30718882.
- ^ Brown, C. Titus; Tiedje, James M .; Howe, Adina; Canino-Koning, Rosangela; Hintze, Arend; Pell, Jason (2012-08-14). „Škálování sestavy sekvence metagenomu pomocí pravděpodobnostních de Bruijnových grafů“. Sborník Národní akademie věd. 109 (33): 13272–13277. arXiv:1112.4193. Bibcode:2012PNAS..10913272P. doi:10.1073 / pnas.1121464109. ISSN 0027-8424. PMC 3421212. PMID 22847406.
- ^ Birol, Inanc; Warren, Rene L .; Coombe, Lauren; Khan, Hamza; Jahesh, Golnaz; Hammond, S. Austin; Yeo, Sarah; Chu, Justin; Mohamadi, Hamid (01.05.2017). „ABySS 2.0: zdrojově efektivní shromažďování velkých genomů pomocí Bloomova filtru“. Výzkum genomu. 27 (5): 768–777. doi:10,1101 / gr.214346.116. ISSN 1088-9051. PMC 5411771. PMID 28232478.
- ^ Chikhi, Rayan; Rizk, Guillaume (16. 9. 2013). „Prostorově efektivní a přesné znázornění de Bruijn grafu na základě Bloomova filtru“. Algoritmy pro molekulární biologii. 8 (1): 22. doi:10.1186/1748-7188-8-22. ISSN 1748-7188. PMC 3848682. PMID 24040893.
- ^ Loman, Nicholas J .; Misra, Raju V .; Dallman, Timothy J .; Constantinidou, Chrystala; Gharbia, Saheer E .; Wain, John; Pallen, Mark J. (květen 2012). "Porovnání výkonu stolních vysoce výkonných sekvenčních platforem". Přírodní biotechnologie. 30 (5): 434–439. doi:10,1038 / nbt.2198. ISSN 1546-1696. PMID 22522955. S2CID 5300923.
- ^ Wang, Xin Victoria; Blades, Natalie; Ding, Jie; Sultana, Razvan; Parmigiani, Giovanni (30. 7. 2012). „Odhad chybovosti sekvenování při krátkých čteních“. BMC bioinformatika. 13: 185. doi:10.1186/1471-2105-13-185. ISSN 1471-2105. PMC 3495688. PMID 22846331.
- ^ Schmidt, Bertil; Schröder, Jan; Liu, Yongchao (01.02.2013). "Musket: vícestupňový k-mer spektrální korektor chyb pro data sekvence Illumina". Bioinformatika. 29 (3): 308–315. doi:10.1093 / bioinformatika / bts690. ISSN 1367-4803. PMID 23202746.
- ^ Hwu, Wen-Mei; Ma, Jian; Chen, Deming; Wu, Xiao-Long; Heo, Yun (2014-05-15). „BLESS: Bloomovooriginální řešení korekce chyb pro čtení s vysokou propustností“. Bioinformatika. 30 (10): 1354–1362. doi:10.1093 / bioinformatika / btu030. ISSN 1367-4803. PMC 6365934. PMID 24451628.
- ^ Pellow, David; Filippova, Darya; Kingsford, Carl (01.06.2017). „Zlepšení výkonu Bloomova filtru na sekvenčních datech pomocí k-mer Bloomových filtrů“. Journal of Computational Biology. 24 (6): 547–557. doi:10.1089 / cmb.2016.0155. ISSN 1066-5277. PMC 5467106. PMID 27828710.
- ^ Zhang, Zhaojun; Wang, Wei (2014-06-15). „RNA-Skim: rychlá metoda kvantifikace RNA-Seq na úrovni transkriptu“. Bioinformatika. 30 (12): i283 – i292. doi:10.1093 / bioinformatika / btu288. ISSN 1367-4803. PMC 4058932. PMID 24931995.